Navigation
Install the app
How to install the app on iOS
Follow along with the video below to see how to install our site as a web app on your home screen.
Note: This feature may not be available in some browsers.
More options
-
By using this site you agree to the terms, rules, and privacy policy.
-
Charlie's Restoration Giveaway #2 (Entire Home EMF Mitigation & Protection Along With Personal Protection) - Click Here To Enter
-
Dear Carnivore Dieters, A Muscle Meat Only Diet is Extremely Healing Because it is a Low "vitamin A" Diet. This is Why it Works so Well...
Rest the rest of this post by clicking here
-
The Forum is transitioning to a subscription-based membership model - Click Here To Read
Click Here if you want to upgrade your account
If you were able to post but cannot do so now, send an email to admin at raypeatforum dot com and include your username and we will fix that right up for you.
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.
You should upgrade or use an alternative browser.
dukesbobby777
Member
- Joined
- Sep 22, 2020
- Messages
- 637
Untrue, salmon has antioxidants like astaxanthin along with the n-3
Vegetable oils and salmon do not have the same issues
That antioxidant stops the n-3 being stored in the tissues when you eat salmon?
The n-3 building in fat storage, and lowering metabolism when FFAs are liberated (due to the increased PUFA load being released into serum), is the long term negative effect from what I understand from Peat. Irrespective of how nutritious salmon is, and what the positive acute effects n-3 have on inflammation (lowering the function of immune system).
ive seen you claim the antioxidant thing multiple times on the forum so im going to give my input.Untrue, salmon has antioxidants like astaxanthin along with the n-3
Vegetable oils and salmon do not have the same issues
Salmon are cold water fish that base most of their life in areas where their PUFA concentration doesnt harm them (though Peat has mentioned that even naturally high PUFA animals would fare better on Sat fat, unless of course they then get dropped in some cold water).
The PUFA keeps the fish mobile in cold temps and the astaxanthin also assists in stability (maybe it has other benefits for the fish im not sure).
This however is much different than a human environment. We physiologically operate and live in high temperature places even in cold climates. Preserving PUFA is not the issue, the issue is its accumulation in the body and the fact that its physiologically mismatched to our composition. The less stable HUFAs (like fish oil) are burned relatively fast (in active persons) so the astaxanthin might assist in mitigating harm there, its the PUFA that gets stored in higher amounts. And as great as Vit E and the like are, they arent eliminating PUFA from the body they are just making it easier for the body to handle them when being mobilized, since they are unstable.
IF you were to have any argument for the benefit of things like salmon, it would be that they contain many thyroid supporting nutrients (of which astaxanthin is not one) like iodine in normal levels, selenium, copper, zinc and B vitamins, with crustaceans having extra benefit do to their low iron and high copper.
Simply put, our bodies, no matter how white, have not actually left the tropics.
Ritchie
Member
- Joined
- Nov 22, 2015
- Messages
- 490
The point of that article, and this thread, is that this is a statement that needs evidence to back it up. And obviously that evidence cannot be rat, mice or other animal studies, or in-vitro. I'm assuming you're aware of how low on the evidence hierarchy spectrum animal studies are? At the very bottom because time and time again, observations in animals have been shown to not only be untrue in humans, but the complete opposite. The reasons for this should be obvious. So, to state what you have here conclusively, the evidence must be in humans. You seem to be making a vague evolutionary argument here, just doesn't really cut it in the context here.the issue is its accumulation in the body and the fact that its physiologically mismatched to our composition
Secondly, you seem to be making the claim that the PUFA you eat is the PUFA you store/wear. Genuinely curious if you have seen any evidence in humans of this? Ie fat composition of stored fat comparing control and experimental groups?
TopGun1911
Member
- Joined
- Aug 7, 2020
- Messages
- 48
Personally, I start to suffer from dry eyes and cognition issues if I go without omega-3 for a long time. Just a can of sardines a week solves this for me.
postman
Member
- Joined
- Mar 3, 2016
- Messages
- 1,284
Keeping PUFA intake below 4 grams per day is definitely enough to upregulate mead acid production by over a thousand percent. There is a study about this but I can't be arsed to find it again right now.It was Ray himself who talked about the 1 gram to forum member @tca300. Whether you believe that or not is a person’s own personal belief I guess. He basically said that in order to prevent it accumulating with age, you really do need to be that extreme. But the fact that he probably doesn’t do that himself, probably means that he recognizes how hard that actually is. If it were easier and more practical, we’d all be doing it. Personally I just can’t live on skimmed milk, low fat cottage cheese, sugar, very small pieces of liver and hydrogenated coconut oil. I get very bad digestion from dairy that has little fat in it. It’s my understanding that your body has to be very deplete in PUFA in order to ramp up mead acid production. And you won’t get that from 4g per day, as tissue levels will always be very slightly increasing as the years pass. But yes, your quality of life will be much better at that amount than neglecting PUFA intake and eating the SAD diet instead (or other health inspired diets that allow for ample PUFA consumption).
For those of us who are less extreme we have things like coconut oil (dilutes PUFA), vitamin E, aspirin and niacinamide which can help against PUFA.
Ray has also said that it takes about 4 years of PUFA avoidance to achieve a depleted state. If you eat a couple of grams of PUFA there might not be 100% depletion but that's a bit of a rediculous goal anyways. You might still be accumulating PUFA in certain specific tissue but the fat in your adipose tissue will reflect the fat that you consume. If you go from your fat being 30% PUFA to it being 2% PUFA, is that not depletion? Because it's not 0%?
postman
Member
- Joined
- Mar 3, 2016
- Messages
- 1,284
They do have the same issues, have you read Ray's articles about fish fat?Untrue, salmon has antioxidants like astaxanthin along with the n-3
Vegetable oils and salmon do not have the same issues
keytothecity
Member
- Joined
- Dec 5, 2020
- Messages
- 204
Correct, and hypothyroidism is cooling your body down to keep pufa more stable because of itive seen you claim the antioxidant thing multiple times on the forum so im going to give my input.
Salmon are cold water fish that base most of their life in areas where their PUFA concentration doesnt harm them (though Peat has mentioned that even naturally high PUFA animals would fare better on Sat fat, unless of course they then get dropped in some cold water).
The PUFA keeps the fish mobile in cold temps and the astaxanthin also assists in stability (maybe it has other benefits for the fish im not sure).
This however is much different than a human environment. We physiologically operate and live in high temperature places even in cold climates. Preserving PUFA is not the issue, the issue is its accumulation in the body and the fact that its physiologically mismatched to our composition. The less stable HUFAs (like fish oil) are burned relatively fast (in active persons) so the astaxanthin might assist in mitigating harm there, its the PUFA that gets stored in higher amounts. And as great as Vit E and the like are, they arent eliminating PUFA from the body they are just making it easier for the body to handle them when being mobilized, since they are unstable.
IF you were to have any argument for the benefit of things like salmon, it would be that they contain many thyroid supporting nutrients (of which astaxanthin is not one) like iodine in normal levels, selenium, copper, zinc and B vitamins, with crustaceans having extra benefit do to their low iron and high copper.
Simply put, our bodies, no matter how white, have not actually left the tropics.
Prevents cancer when pufa is there
ATP
Member
- Joined
- Oct 15, 2015
- Messages
- 279
PUFAs are utilized as substrates for pro-inflammatory action and unsaturated fatty acids are susceptible to lipid peroxidation. Human nutritional studies universally have a reputation of being inaccurate with varying degrees of validity. The author utilizes these studies to conclude that the pro-inflammatory mechanism and unstable nature of highly unsaturated fatty acids do not translate into negative health effects.
Even significantly reducing dietary PUFAs may not be enough to negate the effects of liberated free fatty acids which none of the studies took into consideration.
Even significantly reducing dietary PUFAs may not be enough to negate the effects of liberated free fatty acids which none of the studies took into consideration.
OP
DANIEL
Member
- Joined
- Nov 10, 2020
- Messages
- 77
For many reasons, I think Peat, not Masterjohn, should reconsider his stance on essential fatty acids (I'll just say PUFA hereafter). Peat assumes PUFA not essential because he assumes there is no lipid bi-layer membranes on cells and mitochondria. Scientists like Masterjohn assume the bi-layer exists, and that linoleic acid (the parent omega-6) is a critical component, responsible for the passage of oxygen and other materials through the membrane. Once Peat makes these assumptions (non-essentiality of PUFA, no such thing as membranes) he then proceeds to recommend eliminating PUFA from the diet, or at least minimizing it. He leaves his followers with this blanket rule, yet proceeds to recommend foods that have substantial PUFA content, I guess hoping that no one will check up on him and call him on it. It's hard to maintain a following of careful investigators this way.
It's fine if you ignore the essential nature of omega-6. I won't fear for your life because unless you have a million dollar lab working to refine all the omega-6 out of each food you eat, you're going to get a supply of it, and probably an ample supply. My policy is that fear of PUFA is silly. Now fear of ruined, oxidized PUFA is a different matter. I'm of the impression that the important counsel here is to get your foods as fresh as possible and not let them sit around, age, and oxidize. I think the main thing Peat wants is for people to get off excess, ruined PUFA. Avoidance is impossible. He admits that. So arguing requirement of omega-6, in practicality, is a mute point.
When you say that "in Lipid Lipid Bi-layer theory its stated that the composition of the cell bi layer is dependant on dietary intake" I think you mean to imply that there is a bi layer somewhere that happens to be free of PUFA. I think, if there is such a theory, the reality is that the amount of PUFA varies to some degree, but is not zero in any cell. In fact, it should be simple to find a cell somewhere that has no PUFA. If this has been done, let's find the study. In fact the universal appearance of PUFA in all cells is almost a de-facto proof of it's essential nature. Nature is pragmatic and efficient. Never are you going to find a substance in animals and plants, present in every cell since we've been doing assays, that isn't a part of the chemistry. Just this alone argues quite well against "who says omega 6 is absolutely required?". Why use science when you have statistics?
It seems that the OP wishes to contrast Masterjohn with Peat, and one of the points of contrast is this essentiality issue. That is what I was responding to. I said Peat tended to "recommend eliminating PUFA from the diet, or at least minimizing it." If you havent' read anything from Peat like that, keep reading. He treats PUFA very carelessly in some statements, and only slightly more carefully in other statements, and people misinterpret or fail to look deeper to get clarification. So sometimes he does qualify that it's impossible to avoid PUFA, but sometimes he doesn't, and implies that PUFA should be avoided. That's his problem, and one reason we even have a forum. Vagueness, lack of detail, refusal to clarify, etc. Things that most of us here are careful to do, but Peat has no time or inclination for, apparently.
As to the "zero-fat experiments" you mentioned, I'd be happy to look at them if you provide a link. However, I will say I've checked out about a dozen links on PUFA studies that Peat has cited in the past, and every single one of the studies has proven that there is a problem caused by PUFA by using, not just your grocery store variety refined seed oils, but bulk, commercial seed oils (usually corn or soy). From my point of view, these studies (usually funded by drug companies) were a total waste of money. They prove nothing except that ruined food is bad for health. For the typical person who reads Peat and other alternative gurus, and who is already on a road (hopefully) to better health via intake of only whole, organic, and unprocessed foods, the proof that adulterated oils cause health problems is not even a surprise or of value. Most already know this. The serious issue though that I'd like to clear up here is this: It's just a shame when a writer calls all linoleic acid (omega-6) and all alpha-linolenic acid (omega-3) "PUFA" (including all their derivatives that cascade, on demand, from these parent oils), and refers only to "PUFA" when they talk about the advantages or disadvantages, but never mentions that what is a healthy ingredient in all our foods has been ruined by processing, and that it is the processed and probably oxidized PUFA that is to be avoided. I consider the lack of differentiation between healthy sourced unsaturated fats, and ruined versions of the same omega's to be inexcusable.
But I admit, I have a different view of the effect of PUFA in the body than Peat. He considers the oxidation possibility in the body as the threat, and I trust the body to take care of healthy PUFA, and only see previously, externally oxidized PUFA as the threat. I consider that yes, there is a lipid bi-layer around each cell, and around all the mitochondria, and once PUFA is integrated in that bi-layer, it is safe for a while, and effective at performing a function (thanks to the protecting presence of plentiful saturated fat also in that bi-layer which acts as an anti-oxidant). But whether there is a bi-layer or not, we know that PUFA is an integral part of the cells. And when, for the purpose of health, the body deconstructs a cell, any PUFA which needs to be removed will then be removed. As you are probably aware, all of us developed a very useful brain during the months in-utero and the 2 years after birth. During that time, one of the most vulnerable of the PUFAs (DHA) was included into our brains, in cells which will not be replaced for a lifetime. The entire stock of DHA came from our foods during that time, and much of that remains, unchanged, year after year, because the cells are not replaced. If this can take place in the brain, why cannot PUFA be preserved in relative safety in the cells of other organs? I don't know how this is done, but I assume it's part of our physiology, and has been for millions of years.
BigYellowLemon
Member
- Joined
- Jul 6, 2016
- Messages
- 550
I couldn't quote this normally because of a forum software bug.
"BigYellowLemon said:
Here's what I don't understand about Ray Peat: he's absolutely convinced, justifiably so, that PUFA is completely unnecessary in the diet. And he seems to think that it's the root cause of all aging and pretty much everything bad. And yet, he doesn't eat a PUFA free diet.
You would think he would be the going extreme about it, only consuming hydrogenated fats, but he drinks fatty milk, uses butter, and eats egg yolks.
And sure, those things are good, the best PUFA ratios among the natural goods. But here's the thing, if you want to actually be PUFA-free, if you actually want to deplete your body of PUFA and experience the benefits of their absence, then you have to be extreme. To actually deplete yourself of PUFA, you need to eat less than 1g of it a day, and even that wouldn't be true depletion, but good enough.
This makes fully hydrogenated fats + fat free foods a necessity."
Ray drinks skim milk, and he recommends 1 egg per day, from real free range chicken who eat their own natural non-grain feed diet.
To rev up your production of mead acid you need to stay below 4-5 grams of PUFA per day, not 1 gram. This "you have to eat than 1 gram of PUFA per day" stuff has no basis, the goal isn't to have a body with 0% PUFA. The less PUFA you eat the better off you will be, and the less PUFA you will carry on your body, but there is no perfect food.
It was Ray himself who talked about the 1 gram to forum member @tca300. Whether you believe that or not is a person’s own personal belief I guess. He basically said that in order to prevent it accumulating with age, you really do need to be that extreme. But the fact that he probably doesn’t do that himself, probably means that he recognizes how hard that actually is. If it were easier and more practical, we’d all be doing it. Personally I just can’t live on skimmed milk, low fat cottage cheese, sugar, very small pieces of liver and hydrogenated coconut oil. I get very bad digestion from dairy that has little fat in it. It’s my understanding that your body has to be very deplete in PUFA in order to ramp up mead acid production. And you won’t get that from 4g per day, as tissue levels will always be very slightly increasing as the years pass. But yes, your quality of life will be much better at that amount than neglecting PUFA intake and eating the SAD diet instead (or other health inspired diets that allow for ample PUFA consumption).
For those of us who are less extreme we have things like coconut oil (dilutes PUFA), vitamin E, aspirin and niacinamide which can help against PUFA.
Yes, thank you. Ray himself has said many times that the lower, the better, and that lower than 1g would be best.
Trust me, over the past 4 years I've read and watched nearly everything Ray has said. And he has said in the past that he eats lots of butter and lots of fatty beef, and has drank 1% milk for years, probably decades. Maybe he drinks skim milk now but for a while he drank 1% milk.
You're also wrong about the Mead acid. That requires sub-gram PUFA consumption. 4-5g isn't going to cover it. Even 400mg or so of linoleic acid can shut down Mead acid production.
It's not like Mead acid is anything special though. By definition it is a PUFA itself; it has 3 double bonds, 1 more than linoleic acid. The only real use Mead's acid has is as a potential replacement for DHA, but that's theoretical anyways.
postman
Member
- Joined
- Mar 3, 2016
- Messages
- 1,284
I don't know why I can't quote your posts but the message you posted above is bull****, Daniel.
Ray recommends one (1) egg per day, that one egg has about 0.7 grams of PUFA. And you don't have to eat it, you can get its nutrition from other foods instead. His other basic recommendation is skim milk and fruit and fruit juices, those foods are virtually fat free. Occasional seafood and cheese have slightly more PUFA but still very low unless you eat massive quantities.
In regards to the second post, I don't think he or you understand that when you don't consume PUFA, your body makes its own PUFA, the much more stable omega-9 fatty acids. Mead acid. If you don't eat the toxic omega-3 and omega-6 fatty acids you will instead find omega-9 in your cells.
Ray recommends one (1) egg per day, that one egg has about 0.7 grams of PUFA. And you don't have to eat it, you can get its nutrition from other foods instead. His other basic recommendation is skim milk and fruit and fruit juices, those foods are virtually fat free. Occasional seafood and cheese have slightly more PUFA but still very low unless you eat massive quantities.
In regards to the second post, I don't think he or you understand that when you don't consume PUFA, your body makes its own PUFA, the much more stable omega-9 fatty acids. Mead acid. If you don't eat the toxic omega-3 and omega-6 fatty acids you will instead find omega-9 in your cells.
postman
Member
- Joined
- Mar 3, 2016
- Messages
- 1,284
Below 4g is definitely enough to upregulate mead acid production by A LOT. You will make even more if you eat even less PUFA but if you eat 4g PUFA you will produce more than a 1000% more than if you eat say 15 grams of PUFA.Yes, thank you. Ray himself has said many times that the lower, the better, and that lower than 1g would be best.
Trust me, over the past 4 years I've read and watched nearly everything Ray has said. And he has said in the past that he eats lots of butter and lots of fatty beef, and has drank 1% milk for years, probably decades. Maybe he drinks skim milk now but for a while he drank 1% milk.
You're also wrong about the Mead acid. That requires sub-gram PUFA consumption. 4-5g isn't going to cover it. Even 400mg or so of linoleic acid can shut down Mead acid production.
It's not like Mead acid is anything special though. By definition it is a PUFA itself; it has 3 double bonds, 1 more than linoleic acid. The only real use Mead's acid has is as a potential replacement for DHA, but that's theoretical anyways.
Rafael Lao Wai
Member
- Joined
- Jun 16, 2017
- Messages
- 1,790
Mead acid is PUFA too, that's true, but the difference is the carbon at which the double bonds start. The closer to the carbonyl end the double bond is, the more stable it is. The double bonds in mead acid start only at carbon 9. Contrast that with omega 3s, in which the double bonds start at carbon 3.It's not like Mead acid is anything special though. By definition it is a PUFA itself; it has 3 double bonds, 1 more than linoleic acid. The only real use Mead's acid has is as a potential replacement for DHA, but that's theoretical anyways.
are you implying that 3 and 6 are being produced endogenously?... where else would they come from and how else would yellow fat disease or lipofuscin in humans happen? if observing nature can help one understand complex subjects i dont understand what is wrong with referring to them as evidence. Tropics SaFa, Cold climates PUFA... observe why and thats pretty big evidence right there.The point of that article, and this thread, is that this is a statement that needs evidence to back it up. And obviously that evidence cannot be rat, mice or other animal studies, or in-vitro. I'm assuming you're aware of how low on the evidence hierarchy spectrum animal studies are? At the very bottom because time and time again, observations in animals have been shown to not only be untrue in humans, but the complete opposite. The reasons for this should be obvious. So, to state what you have here conclusively, the evidence must be in humans. You seem to be making a vague evolutionary argument here, just doesn't really cut it in the context here.
Secondly, you seem to be making the claim that the PUFA you eat is the PUFA you store/wear. Genuinely curious if you have seen any evidence in humans of this? Ie fat composition of stored fat comparing control and experimental groups?
Humans knew what to and not to eat for much longer than weve been (poorly) dissecting it "scientifically." I have yet to find any ancient culture that willingly gorged themselves on natural PUFA laden food and A.) didnt suffer consequences or B.) develop some sort of genetic change like the Inuit (refer to Chris Masterjohn Inuit Keto info).
Even if PUFA is in some way "essential" there is no shortage both in and out of the modern body except with great effort and difficulty. So to claim one has a condition because of PUFA deficiency is very myopic and a failure to read Peats extensive material and understand it in context.
Capt Nirvana
Member
- Joined
- Mar 25, 2018
- Messages
- 108
Peat's claim about PUFA is arguably his most controversial viewpoint. A lot of the disagreement has to do with Peat basing his perspective on rat studies and then claiming that his findings apply to humans as well. I came across this pro-PUFA article and I was wondering if you guys could share your thoughts on it and possibly debunk its claims. The author has a Master of Science degree in nutritional medicine.
Article:
Of Rats and Sydney Diet-Heart: Drawing a Line Under Polyunsaturated Pseudoscience
"To say that hysteria about the health effects of polyunsaturated fats [PUFA] has reached fever pitch may be an understatement.
It can be interesting watch a trend of thought gain traction when that train of thought is inconsistent with a substantial total body of evidence, from multiple converging lines of inquiry.
It speaks to something at play beyond scientific discourse, and frankly much of the “debate” regarding PUFA is steeped in a mix of narratives, from fantasies about our evolutionary past to conspiracy theories about governments, scientists, the food industry, and ‘BigPharma’.
I have a few thoughts as to why the PUFA-hysteria appears to have exponentially increased in recent years. It’s likely not an exhaustive list, but these are some of the more common narratives I’ve observed.
1) The surge in popularity of “ancestral” viewpoints of health, and the popularity of ‘Paleo’ diets and other arguments based on evolution and “natural fats”, which apparently means saturated animal fat.
The problem with this is it takes a peculiarly Eurocentric view of evolution, as if our genus evolved somewhere in Scandinavia huddled in bearskins around a slain wooly mammoth, rather than out of the African Rift Valley. The idea that our diet was rich in animal fat is an evolutionary fantasy dreamed up by modern wannabe-cavemen, unsupported by any real evidence. Saturated fat are estimated to have provided about 6% of the average total energy intake for humans in the Paleolithic period, reflecting the fat composition of wild game meat in free-living African mammals. And over half – up to 60% – of the brain’s dry weight is lipid (fat), and of that up to 30% is comprised of polyunsaturated essential fatty acids. Docosahexaenoic acid [DHA] alone comprises over 90% of omega-3 fats acid in the brain, generating a “shore-based perspective” of human evolution.
Stable isotope analysis of bones of early Sapiens have indicated that marine protein sources constituted significant proportions of daily energy in the human diet. Anthropological evidence for the expansion of the genus Homo from the African rift valley suggests a migration along coastal and inland watercourses, provided access to both marine and freshwater sources of fish, and in particular long-chain omega-3 fatty acids. The evidence suggests that the consistent access to such food sources coincided with the period of exponential encephalisation, preceding the rapid development of language, complex reasoning, and problem solving cognitive capacities associated with the prefrontal cortex. Relative to other mammalian species, and indeed our primate cousins, the human brain is disproportionately large compared to body size. Other mammals with high levels of other polyunsaturated fats in membranes, but without a direct dietary source of preformed DHA, did not develop large brains, indicating a foundational need for preformed DHA in the early modern human diet evident in the preference for DHA and polyunsaturation in neural membranes.
2) Ability to find rodent studies to corroborate beliefs about PUFA.
Sure. It is completely uncontroversial that in many rodent studies, lipid peroxidation has been demonstrated relative to the degree of unsaturation of fats in the experimental feed, and the antioxidant capacity of the host animal. It’s odd that such studies in our rodent friends are used to support theoretical arguments of human evolution harking back to the Palaeolithic period. To say nothing of their lack of translational evidence in humans.
3) Play on ‘BigFood-BigPharma-wants-to-make-you-sick’ emotive narratives.
I’m not going to defend the food industry and the role it has played in altering the food environment and deliberately propagating both bad science and a bad food supply. But that argument can still be made grounded in facts and evidence, not emotively-driven conspiratorial beliefs. The idea that PUFA have been injected into our food supply to contribute to a slow and painful demise is nonsense rhetoric. This also tends to run with the “it all started with Ancel Keys” diatribe, regurgitated by automatons who haven’t even read his research, or even of the wider diet-heart literature, from the period. I’ve written an extensive article on the early metabolic ward studies from that period, and the various research groups involved, which you can read elsewhere on site.
4) It’s easy to build a pseudoscientific case against PUFA.
This is most assisted by citing rodent studies without attempting to build a cohesive case or argument. These arguments may seem impressive to the unsuspecting reader. They’re often referenced, which provides a veneer of evidence-based. But closer scrutiny and it’s just a collection of standalone claims with a rodent reference from 50yrs ago at the end. “PUFA have been shown to do X (Rat et al., 1972)”. “Y increases when PUFA are fed (Mice et al., 1965)”. No attempt to cogently synthesise the arguments and look for converging lines of evidence. Then state potential mechanisms as if they’re established in vivo effects, followed by some dazzling with chemistry language as if translational research doesn’t exist. And not to mention, flat out ignore an entire of body evidence of outcomes in humans. Sprinkle on top the usual conspiratorial icing, and it all sounds rather impressive, but the argument is a house of cards.
In addition to the 50yr old rats, we’ve had a resurgence of some dusty old human studies which add to the pseudoscientific veneer: Sydney Diet-Heart and the Minnesota Coronary Experiment in particular like to do the rounds. Citing studies like these involves either total cognitive dissonance or lack of scientific literacy, both of which are mainstream issues within the ‘carnivore’ community and other low-carb iterations. Recently, I’ve also seen reference to the Women’s Health Initiative and the Helsinki Businessman’s Study: we’ll address these briefly when we discuss these studies further, below. Oh, and of course you can also do a meta-analysis with some of these studies, and just like the bad data that goes in, bad data comes out.
The Claims
The gravamen of the argument for PUFA being potentially deleterious for human health centres on two processes: inflammation and oxidation, specifically, lipid peroxidation. In a nutshell, the argument centres on the chemical composition of PUFA resulting in these fatty acids with double-bonds being prone to lipid peroxidation, and therefore adverse effects through oxidative stress. Additionally, that the omega-6 linoleic acid acts as a precursor to pro-inflammatory eicosanoids, and therefore that PUFA cause adverse effects through inflammation.
We’ll look at human evidence in relation to both, but first, let’s discuss a concept that is critical to understand when assessing claims about diet and health: putative mechanism does not equal certain effect.
However, this assumption between putative mechanism – or basic chemistry – and assumed effect underpins much of the anti-PUFA rhetoric. This assumption relies on a rhetorical tactic where a statement of fact is offered as evidence of a conclusion. Let’s take an example of when Cholesterol Denialists argue “but the body NEEDS cholesterol, bro!” and utter some ***t about ‘building blocks’, as if this fact provides evidence of a conclusion that we need high levels of cholesterol. But the science-based reality is that very little amounts are physiologically normal, and we really don’t need that much.
The exact same rhetorical hack is deployed with much of the claims regarding PUFA. “Well you see they’ve a double-carbon bond at the omega-6 position” as if this provides evidence of a conclusion of disease risk in humans, or “well you see linoleic acid is a precursor to arachidonic acid, and that is pro-inflammatory”, as if this provides evidence of an actual inflammatory effect. What they are not telling you, or asking, is the answer to the fundamental question: is this physiologically relevant in humans? Does this putative mechanism translate to an in vivo effect?
Again, from the perspective of the lay person these types of arguments can sound scientific and convincing. But let’s deal with the fundamental question: what is the evidence for these processes in humans as a result of PUFA intake, and are these results relevant?
Inflammation and Lipid Peroxidation (In Humans)
Let’s start with inflammation. The putative mechanism with regard to inflammation is that the omega-6 linoleic acid [LA] acts as a precursor to arachidonic acid [AA]; AA acts a substrate to form eicosanoids, and the eicosanoids AA is used to form may be pro-inflammatory. Ergo, as the logic goes, increasing PUFA – particularly LA – increases inflammation.
Except there is no evidence that either increasing or decreasing LA levels alters levels of AA in humans. A review of 36 human intervention studies highlighted that neither increasing LA levels by up to 551%, or decreasing LA levels by 90%, altered concentrations of AA in plasma, serum, or red blood cells [erythrocytes], despite increasing LA intake resulting in increased membrane phospholipid LA content. Putative mechanism does not = biological effect. The following illustration helps to illustrate why:
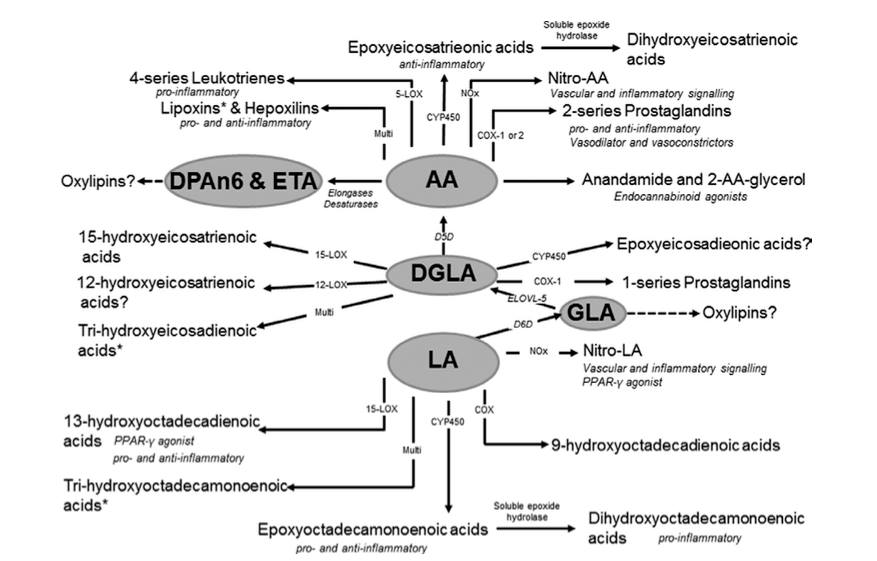
The delta-6-desaturase [D6D] enzyme responsible for converting dietary essential fatty acids into longer-chain fatty acids, is also necessary for conversion of LA to AA, and in adulthood there is very little activity of D6D. As you can see from the above figure, LA is also not directly metabolised to AA, but goes through multiple intermediate conversion processes, which require D6D. The net effect is that AA levels are kept at a relatively constant level in the body, and this is also observed during breastfeeding, where milk is maintained with a constant AA level through triglyceride lipolysis, despite dietary intake.
That is the first step in the putative mechanism of inflammatory effects of LA – conversion to AA – for which there is no evidence of dramatic changes in LA dietary intake altering circulating levels of AA. What about inflammation itself? Johnson & Fritsche conducted a review of 15 human intervention trials investigating the effects of LA on inflammatory biomarkers. Their conclusion was that “virtually no data are available from randomised, controlled intervention studies among healthy, non-infant human beings to show that the addition of LA to diets increases markers of inflammation.”
This is where the buck just stops for the inflammation argument. There is no evidence that the putative mechanism is operative, nor that there is an inflammatory effect of dietary LA in actual Homo Sapiens. And, let’s not forgot both the anti-inflammatory effect of alpha-linolenic acid [ALA] and the anti-inflammatory role of EPA and DHA.
So, what of lipid peroxidation? A search of the terms “polyunsaturated” and “lipid peroxidation” and “human” in PubMed reveals a small number of trials that have directly tested the effects of PUFA on various markers or surrogate markers of lipid peroxidation in our very own species.

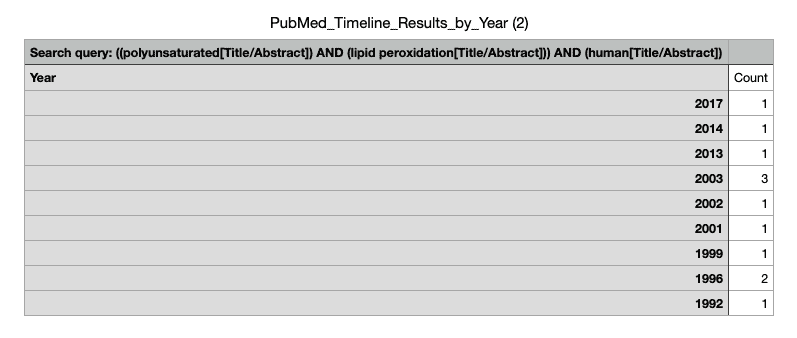
The studies vary greatly in terms of methodology, intervention, and most importantly, the assessment of lipid peroxidation, so a meta-analysis would be pointless. Instead, I’ve qualitatively synthesised the studies. To reiterate: these are studies in actual humans.
Strapans et al., 1994: 6 healthy males were tested in an acute, single meal postprandial study. The control diet was corn oil that was unoxidised plus high vitamin E, compared to two diets with moderate and high levels, respectively, of oxidised lipids together with low vitamin E. These diets contained ~57-59% LA. The results demonstrated that levels of oxidised lipids from the pre-oxidised oils determined levels of oxidised lipids in postprandial chylomicrons. High LA unoxidised corn oil with high vitamin E content (0.14mg per gram oil) had no effect on any of the measured markers of lipid peroxidation. While this study indicates that oxidised oils may be absorbed from dietary intake, the low vitamin E content of the oxidised lipid diets likely biased the results, as vitamin E is an important modifier of lipid peroxidation. More recent evidence suggests consumption of oxidised fish oils does not increase more reliable markers of oxidative stress. Further, the use of indirect assays to measure lipid peroxidation, including thiobarbituric acid reactive substances (TBARS) and conjugated dienes, which are non-specific and may measure products other than lipid peroxidation, and be elevated without concomitant elevations in markers of oxidative stress.
Turpeinen et al., 1995: This study conducted two separated studies, using both in vivo and in vitro analyses. Both studies compared a sunflower oil diet to a rapeseed oil diet. In study 1, 59 healthy adults underwent strict dietary control with a sunflower oil diet containing 13% PUFA [12% LA] compared to a rapeseed oil diet containing 7% PUFA, 5% LA, and 2% alpha-linolenic acid [ALA], against a high saturated fat milk-fat based diet. The milk-based diet was followed for 14-days, followed by 24-days on the sunflower oil diet then rapeseed oil diet for a further 24-days. Half of the participants completed the study in reverse diet order. There were no significant differences in malondialdehyde (MDA) levels, an end-product of lipid peroxidation, or indirect lipid peroxidation assays, including TBARS or conjugated dienes. In the second study, 13 healthy adult participants were assigned to the sunflower oil or rapeseed oil diet to follow free-living for 6-weeks. In this study, levels of TBARS and lipid peroxidation measured in lipoproteins tended to decrease in the sunflower oil diet, although this was only statistically significant for LDL, and exhibit no change in the rapeseed oil diet. However, the in vitro analysis revealed the opposite effect: the PUFA-rich sunflower oil resulted in increased oxidation. This is an important finding because it demonstrates that oxidation may be demonstrated in vitro, which does not correspond with measured in vivo effects. This could have been attributable to the high vitamin E content of the diets, particularly the sunflower oil PUFA diet, with the implication that antioxidant availability in vivo is a more relevant determinant of potential lipid peroxidation.
Jenkinson et al., 1999: This was a free-living study in 10 healthy males comparing diets with 5% PUFA to 15% PUFA, with carbohydrates, fats, protein, and vitamin E matched. Again, this study used outcome markers that are not reliable measures of oxidative stress, in particular TBARS. The authors themselves highlight the non-specific nature of this assay, and the fact that it may measure byproducts that do not result from lipid peroxidation. The study did find that oxidised glutathione was higher on the 15% PUFA diet, however, there was no difference in total or reduced glutathione, or red blood cell glutathione. The finding in relation to oxidised glutathione may reflect an insensitive measure used in this study. Concentrations of oxidised glutathione are very low, and changes may not necessarily reflect oxidative stress. Accurately measuring oxidised glutathione requires high-performance liquid chromatography, which was not conducted.
Södergren et al., 2001: This was a free-living study in 19 healthy adults with diets provided to participants, comparing a rapeseed oil enriched diet to a saturated fat enriched diet. Each diet was consumed for 4-weeks, separated by a 4-week washout period, with participants randomised to order of intervention. The study used reliable measures of in vivo oxidative stress, specifically isoprostanes, in addition to malondialdehyde (MDA). There were no significant difference in markers of oxidative stress or lipid peroxidation, while circulating levels of g-tocopherol increased significantly during the rapeseed oil diet, suggesting the levels of antioxidant naturally present in the oil is sufficient for protective effects.
Nielsen et al., 2002: This was a controlled study in 18 healthy males measuring in vitro effects of three diets enriched with rapeseed oil, olive oil, or sunflower oil, respectively. Compared to the olive and rapeseed oil diets, the sunflower oil diet increased susceptibility of VLDL and LDL to oxidation. However, this was in vitro, and this effect of sunflower oil in vitro was also observed in the study above by Turpeinen et al, who found the opposite, i.e., no effect in vivo.
de Kok et al., 2003: This was a supplemental intervention in 30 healthy females comparing two levels of LA intake, 15g/d and 7.5g/d, to 15g/d palmitic acid, taken for 6-weeks. MDA and markers of oxidative DNA damage were the primary outcomes, and there was no increase in any marker of oxidative stress or DNA damage at any level of LA intake.
Egert et al., 2007: This was a controlled dietary intervention in 75 young adults comparing 3-weeks on rapeseed oil diets enriched with ALA, EPA, and DHA, respectively, after controlled 2-week matched diet run-ins. The study oils contained 7.5% omega-6 and 2.5% omega-3 PUFA, and the oils contained high vitamin E content. Ex vivo oxidation of LDL was the primary outcome. ALA did not effect LDL susceptibility to oxidation, while the effects of EPA and DHA were inconsistent. LDL susceptibility to oxidation was greatest during the DHA-enriched diet. To reiterate a theme, this study is yet another example of the need for caution in extrapolating results from in vitro or ex vivo models to an in vivo effect, and the need to ask whether the finding could be physiologically relevant in vivo. DHA and EPA may have had, albeit inconsistently, effects on LDL susceptibility to oxidation ex vivo in this study: reconcile that against the plethora of data from prospective cohort studies, tissue biomarker studies, and intervention studies, demonstrating the benefit of these PUFA for cardiovascular and neurodegenerative disease.
Freese et al., 2007: This was a feeding study in 77 healthy adults with diets controlled for 6-weeks, comparing diets of 11% and 3% LA for effects on lipid peroxidation and oxidative DNA damage. Isoprostanes measured oxidative stress, and the study measured an in vivo nucleotide which reflects oxidative DNA damage. MDA, TBARS, and ex vivo measures of LDL susceptibility to oxidation were also analysed. Overall, there were no significant differences in any marker of oxidative stress, lipid peroxidation, or oxidative DNA damage.
The foregoing studies are interventions which have directly tested the effects of PUFA, either in diet or supplements, on markers of lipid peroxidation and/or oxidative stress. It is important to be sensitive to method, as many studies have used indirect or non-specific assays, which may not in fact measure lipid peroxidation or reflect oxidative stress. More recent studies using isoprostanes, which have strong utility as biomarkers of oxidative stress, have not demonstrated oxidative effects of PUFA in vivo. Arguably the most important overall finding to synthesise from those studies is the difference between in vitro or ex vivo models, and actual in vivo effects. The disconnect reflects the difficulty with extrapolating results from in vitro studies to assumed effects in vivo, and collectively it appears that there is little evidence from controlled intervention studies that lipid peroxidation from PUFA intake, of various total PUFA intake and varying LA levels, is a major cause for concern in vivo. This is a crucial point in the interpretation and representation of the evidence regarding PUFA.
With the inflammation and oxidation claims addressed, let’s turn to the oft-cited studies proffered as evidence from interventions in humans of an adverse effect of PUFA.
The Oft-Cited Human Studies
Most people never would have heard of the Sydney Diet Heart Study or the Minnesota Coronary Experiment likely if not for Dr Christopher Ramsden, who conducted a re-analysis of both studies, and a meta-analysis with these studies and other studies which used LA interventions, like the 1965 Rose et al. Corn Oil Trial. Ramsden is now celebrated as a demi-god within low-carb/ancestral/paleo/carnivore circles for apparently uncovering “tHe tRUth”. I’m not laying any blame at Ramsden’s door, for the record, merely stating that “reanalysing” old, flawed studies with methodological shortcomings doesn’t make them better studies. You can’t put lipstick on a pig, and you certainly can’t take a pig, donkey, and giraffe and attempt to put lipstick on them together in a meta-analysis.
Let’s start with Sydney Diet-Heart Study [SDHS]. The SDHS was a randomised controlled trial conducted between 1966 to 1973 in 458 men aged 30-59 years who had suffered a recent coronary event (myocardial infarction, acute coronary insufficiency, or angina). Baseline SFA intake was ~16% and PUFA ~6%; the intervention group were advised to increase PUFA intake to 15% energy by replacing SFA [lowering SFA to <10%]. The intervention was provided to participants in the form of a safflower oil and safflower margarine. Median follow-up time was 3.3yrs, the original study only reported on all-cause mortality, for which there was a statistically insignificant increase in risk [HR 1.62, 95% CI 1.00-2.6]). The Ramsden reanalysis published results for cardiovascular disease, indicating statistically significant increased risk for CVD mortality [HR 1.70, 95% CI 1.03-2.80] and coronary heart disease mortality [HR 1.74, 95% CI 1.04-2.9]. The confidence intervals are, however, so wide as to indicate weak statistical power and render difficulty inferring where a true effect may lie.
But let’s focus on the major problems with the SDHS. First, two distinct intervention foods were provided: an oil and a margarine. We have no data on whether participants consumed more of one or the other, but the increased mortality in the intervention group lends a clue. Hydrogenated fats added to oils to make commercial margarines at the time contained up to 25-40% of fatty acids as trans-fats. The margarine used in the study, Marrickville Margarine from the brand ‘Miracle’, was a safflower oil-based margarine produced for commercial use. And the intervention group substantially increased PUFA intake with the provided foods, likely consuming substantial amounts of trans-fats – which are unequivocally associated with significant increased risk for CVD. Moreover, attributing this effect to LA is not possible, because while the oil may have had a high LA content, margarines at the time were predominantly monounsaturated fats, together with the high trans-fat content. There are so many unknown variables with this study in terms of the exact fatty acid composition of the diet, other than the broad label of ‘PUFA’, which is likely an inaccurate description of diet in the first place given the aforementioned factors. And the bottom line is that no one is in a position to dispel the potential confounding. In that regard, SDHS simply shouldn’t be cited, when better quality more recent data exists.
So how do we reconcile it? Against the wider evidence-base, of course. We can start with a meta-analysis from 2014 which conducted a subgroup analysis of 8 omega-6 LA intervention trials, finding no significant effect including the SHDS: removing the SDHS in a sensitivity analysis resulted in a 21% reduction in CVD risk from LA intake [HR 0.81, 95% CI 0.68-0.98]. But we’ll save the rest of the evidence base for the next section, first let’s put a fork in the Minnesota Coronary Experiment [MCE].
Not satisfied with dredging up the SDHS, Ramsden et al. went looking for more data to dust off and ‘reanalyse’. The reanalysis, published in the BMJ (obviously, anything that runs with heart disease revisionism is a lock), was of another intervention conducted in the mid 1960’s in 9,057 patients attending state psychiatric hospitals in Minnesota. The control diet was a diet high in saturated fat, compared to the intervention diet designed to be high in PUFA, and study meals were provided to patients while they were in the institution. The outcomes were blood cholesterol levels and coronary heart disease events. In the original study, there was a statistically insignificant 8% [HR 1.08, 95% CI 0.84-1.37] relative risk increase in risk for coronary events in the intervention group. The confidence intervals in this study are spread across the null so as to make the results practically meaningless to interpretation. The Ramsden et al. re-analysis looked at the associations between blood cholesterol levels and risk of CHD mortality, in 2,355 participants with baseline and follow-up cholesterol measurements. This reanalysis suggested that a decrease in cholesterol of 0.78mmol/L [30mg/dL] was associated with a statistically significant 22% [HR 1.22, 95% CI 1.14-1.32] relative risk increase of all-cause mortality, only in participants aged over 65yrs, irrespective of which diet they were assigned to.
Here is the thing: MCE was a poorly executed study by any standards. Participants were not exclusively in-patients; many were in and out of hospitals, meaning that their participation in the intervention was not continuous, and they consumed study meals while in hospital, before returning to their day-to-day lives. For these participants who were in and out of hospital, the cholesterol measurements were taken after 3-weeks back on the diet following reentry. Without knowing what their levels were upon reentry to the hospital, it is possible that the period after reentry influenced the results. Let’s be really clear about what the MCE, and the reanalysis of the cholesterol levels, can support: nothing. It is incorrect to state that the MCE is evidence of an increase in risk from PUFA, because the original trial findings were insignificant and spread across the null. It is incorrect to state the the PUFA intervention resulted in lower cholesterol and increased mortality risk, because the same effect was observed in the high SFA group. And, finally, the fact that, irrespective of diet, the risk of increased mortality risk with lower cholesterol levels was only in the >65yo participants implies what Rose and Shipley termed the “unsuspected sickness phenomenon”, characterised by an underlying disease resulting in lowering cholesterol levels as the metabolic consequence of the disease. This is a common occurrence in this age group, an observation that allows some to play statistical gymnastics trying to show that higher cholesterol is associated with “longevity”.
Time out for a quote:
This quote by Professor Sander Greenland is referring to meta-analysis, and this is apt for the next study because, as if the reanalysis of SDHS and MCE wasn’t enough, Ramsden et al. conducted a meta-analysis with these two trials and one more, the Corn Oil trial from 1965. And to reaffirm the quote above, a meta-analysis does not solve any fundamental problems in the input data; if the input data is bad, the output is worse. As stated, the relative risk and confidence intervals in MCE render the interpretation of the study meaningless, while the Corn Oil trial resulted in a relative risk of 4.64 [95% CI 0.62-37.15]. The overall meta-analysis of these trials was insignificant, and the near-significant association was based largely on the direction of effect in the SHDS reanalysis, and Rose Corn Oil trial. One has to beg the question, with the preponderance of data available from multiple, converging lines of evidence, what purpose a meta-analysis of three studies from the mid-1960’s with the limitations identified above served. Other than bias confirmation for the heart disease revisionists.
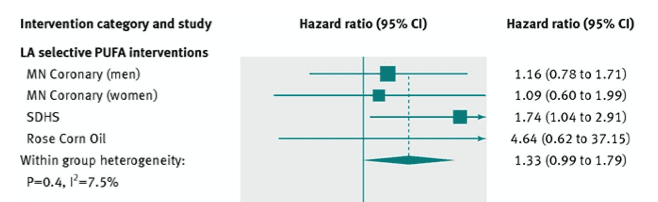
Forest plot from Ramsden et al. meta-analysis illustrating barn-door confidence intervals, low statistical power, and effects of incorporating flawed input data on GIGO [garbage in, garbage out].
The final two studies to be briefly mentioned, which I’ve recently seen added to the SDHS and MCE as proof that replacing SFA with PUFA apparently increases risk, are the Women’s Health Initiative and the Helsinki Businessman’s Study. In the WHI, participants in the intervention group were counselled to reduce saturated fat to ~8% energy, and increase wholegrains, fruit and vegetable, and PUFA intake. While the intervention group managed to reduce SFA from 12.7% to 9.5% over 6yrs, nothing else was achieved for the intervention diet. PUFA decreased from 7.8% to 6.1%, and carbohydrates from refined sources increased. The lack of effect of lowering SFA is explainable by the replacement with largely refined carbohydrate, as evident in the wider literature. So the WHI is categorically not evidence that increasing PUFA has any adverse outcome. Finally, the Helsinki Businessman Study. The 5-year multifactorial intervention in men with elevated cardio-metabolic risk factors advised participants to replace SFA, cholesterol, sugar, with PUFA (mainly as soft margarine), fish, chicken, and vegetables, in addition to physical activity, medications, and smoking cessation. Despite significant reductions in risk factors, incidence of coronary events tended to be higher in the intervention group than in the control group – 19 vs. 9 cases, respectively. However, the analysis of the study indicates that the increase in mortality risk had nothing to do with diet. Univariate and multivariate analyses indicated that the increased risk in the intervention group was associated with use of beta-blockers and clofibrate. Incidence of CHD was lower in the groups who used diet only.
Ultimately, we are talking about a handful of human intervention studies from 60yrs ago which purport to find a particularly detrimental effect of PUFA, and LA in particular. They are methodologically flawed, mostly statistically insignificant, infer no confidence in the statistical power of the findings, and have critical variables – like trans-fats – which can never fully be accounted for. But even if we assumed for a minute that they were sound studies, they would still be a handful of studies in the context of a wider literature accumulated from multiple lines of inquiry, from metabolic wards to prospective cohorts to RCTs, which all converge to support a benefit to PUFA. That is how conclusions are derived in science; not from a rat, a single study, or a handful of studies, but an accumulated total body of evidence.
Converging Lines of Evidence (In Humans)
Ecological data. Prospective cohort studies. Tissue biomarker studies. Metabolic ward studies. Intervention trials. Meta-analyses. In humans.
Metabolic Studies:
This body of evidence dates back to the tightly controlled metabolic ward studies in the 1950’s conducted by various research groups at the time. This period is also important, because within the scientific community there was already evidence that industrial hydrogenation might alter the physically properties of a fat or oil, in turn changing it’s physiological effects. Here is an example of this from Bronte-Stewart et al., published in The Lancet in 1956, which indicated the difference in blood cholesterol levels in response to groundnut oil vs. hydrogenated groundnut oil:
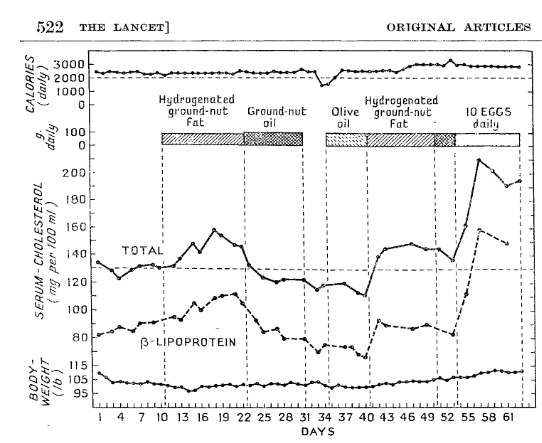
Figure from Bronte-Stewart et al. illustrating the effects on blood cholesterol and body weight of a controlled feeding metabolic ward study manipulating fat and dietary cholesterol sources from hydrogenated groundnut oil, groundnut oil, olive oil, and eggs; clearly differential effects can be seen between the effects of hydrogenation.
This effect of hydrogenation was evident in a number of studies from these various research groups at the time, highlighted in the following quote from a review from Page et al. published in JAMA in 1957:
Thus, the effects of hydrogenation of vegetable oils was evident early in this literature. As we know, however, unfortunately this did not translate into food industry practices for decades.
Keys et al. spent six years experimenting with controlled diets in metabolic studies, varying only in the type and quantity of dietary fat [from 9-44% energy], which ultimately resulted in the ‘Keys Equation’, a predictive equation for the impacts of saturated, monounsaturated, and polyunsaturated fats, and dietary cholesterol, on blood cholesterol levels. The Keys Equation remains valid today. The most important contribution from the Keys Equation was identification of the ‘P:S ratio’, i.e, the ratio of polyunsaturated to saturated fats in the diet, as the most important factor determining blood cholesterol levels in response to diet. To quote from Keys et al. in The Lancet in 1957:
Good science is reproducible, so let’s jump forward by several decades to two meta-analyses of nearly 400 metabolic ward studies, Hegsted et al., 1993, and Clarke et al., 1997. Hegsted et al. confirmed the validity of the Keys Equation in a regression analysis modelling the effects of dietary fatty acids and cholesterol on blood cholesterol levels, in a synthesis of metabolic ward studies published up until 1991. To quote:
Clarke et al. published a meta-analysis of 390 metabolic ward studies modelling both univariate effects [i.e., each dietary constituent in isolation], and multivariate effects [i.e., the effects of substitution/relationship between dietary constituents], demonstrating unequivocally that the effects of isocaloric replacement of energy from SFA with PUFA has the greatest impact on reducing LDL-cholesterol, triglycerides, and overall blood lipid profiles.
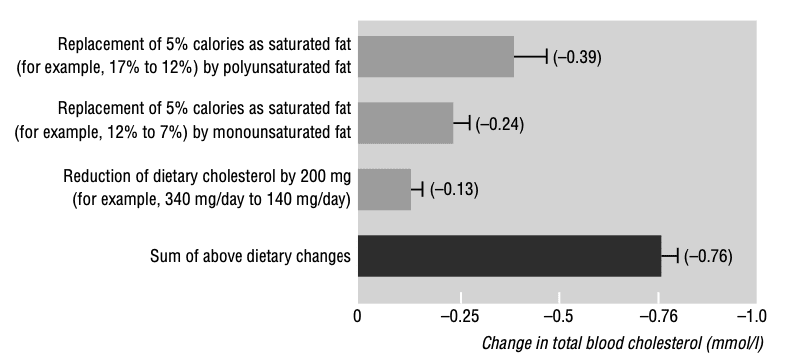
Figure from Clarke et al. demonstrating the effects of isocaloric replacement of saturated fat with unsaturated fats, and reduction in dietary cholesterol, on blood cholesterol. A clear hierarchy is evident with polyunsaturated fats exerting the greatest lipid-lowering effect.
As short-term metabolic ward studies conducted over periods of 2-8 weeks, these studies are looking specifically at intermediate risk factors. In this regard, they provide mechanistic plausibility to the effects on cardiovascular disease reduction from the replacement of SFA with PUFA evident in long-term prospective studies.
Ecological Studies:
It doesn’t necessarily take controlled feeding studies to observe the effects of replacement in the diet. There have been some interesting ecological observations occurring as a result of political upheaval, and also population-wide public health initiatives. Let’s start with the observations from Eastern Europe following the fall of the Soviet Union, in which certain countries experienced a rapid decline in CHD/CVD rates, attributed to a period of market transformation and expanding of the food supply. This sharp decline in heart disease mortality was correlated with a significant increase in population intake of the PUFA, alpha-linolenic acid [ALA]. Traditionally in Eastern Europe, rapeseed or sunflower oil were the predominant oils, but rapeseed oil – rich in both monounsaturated omega-9 and polyunsaturated omega-3 – had been more expensive under Communist regimes, and thus animal fats or sunflower oil, which is low in ALA, remained the predominant cooking fats and oils. Market transformation made rapeseed oil more accessible, and its consumption increased significantly in Poland, Latvia, Lithuania, Czech Republic, and Slovakia: these countries with the greatest increase in grams per day of ALA showed the greatest reduction in mortality. In contrast, Russia, Bulgaria and Romania, which showed no increase in ALA, had no change in CVD mortality.
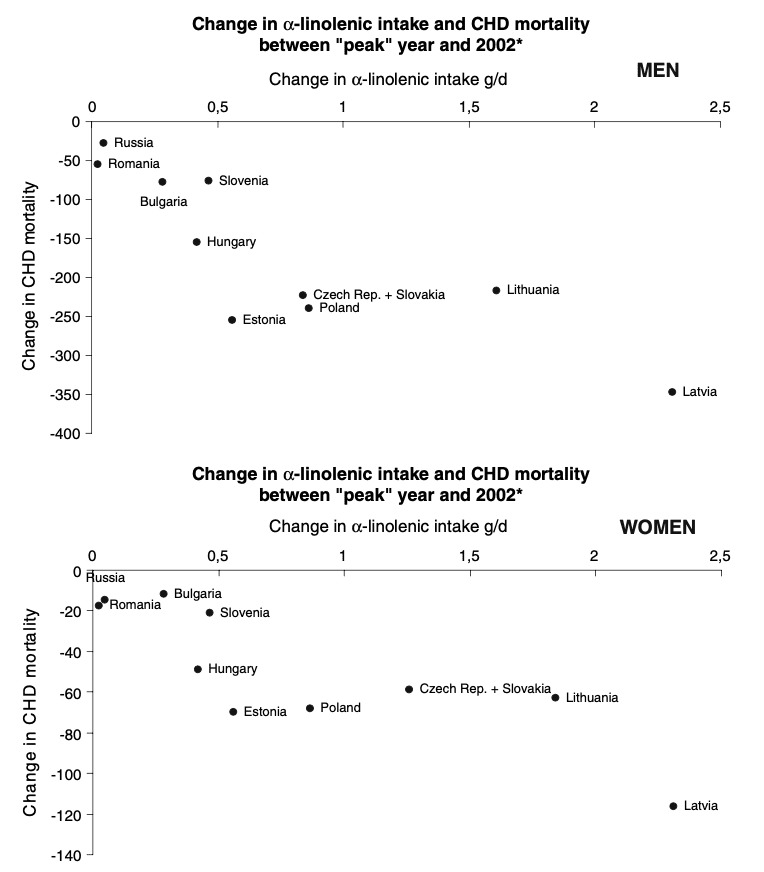
Figure from Zatonski et al. depicting changes in ALA intake and associated changes in CHD mortality in former Soviet countries after 1991.
While the observation in Eastern Europe resulted from external and unforeseen political forces, starting in the early 1970’s Finland embarked on a targeted public health campaign to reduce CHD/CVD mortality. In the 1960’s Finland had the highest population SFA intake in the world at 21-23% energy, with the highest population blood cholesterol levels, and experienced the highest CHD/CVD mortality globally. Starting in 1972, a public health intervention to reduce mortality targeted population reductions in specific risk factors, in particular smoking rates, blood cholesterol, blood pressure, and adiposity. By 2007, CHD/CVD mortality had declined by 80% and of the cumulative risk factors, population reductions in blood cholesterol accounted for 67% of the decrease in mortality. A primary determinant in the population-wide reduction in blood cholesterol was a reduction in dietary saturated fat from 21-23% to 12-13%, achieved through deliberate public health messaging regarding butter consumption [the most significant contributor to SFA in Finnish diets at the time], with dietary advice to replace SFA with PUFA [amongst other dietary advice, including increased vegetable intake and lean meats over fatty meats]. Of particular note, this decrease occurred in the context of smoking rates remaining largely similar [slight decline in men, increase in women], and an overall increase in BMI across the population, both significant risk factors for CVD.
These are ecological studies and population-wide observations, so the usual caveats apply. The Finnish example is also not wholly attributable to PUFA, given that SFA have twice the cholesterol-raising effect as PUFA have lowering cholesterol, i.e., the reduction in SFA across the population was the strongest determinant of reducing population-wide cholesterol levels, and heart disease mortality. Nonetheless, these ecological observations are congruent with the wider literature, both prospective and interventional, and thus have value in the synthesis of converging lines of evidence.
Prospective Cohorts:
In a 23-year mortality follow-up of the Israeli Ischemic Heart Disease Study in 10,059 male civil servants published in 1993, using LA to determine the P:S ratio indicated that the greater the P:S ratio, i.e., the greater the ratio of LA to SFA, was associated with reduced CHD mortality, as was LA alone as a percentage of total fat intake [confidence intervals were not shown]. Further, adjustment for blood cholesterol levels render the findings in relation to dietary fat null, indicating that the relationship between the P:S ratio and CHD mortality was mediated by blood cholesterol levels, as would be predicted from metabolic ward studies.
In 2009, Jakobsen et al. published an analysis of data pooled* from 11 cohorts, including 344,696 participants with a range of from 1.7% to 10.6% PUFA intake 9.4-21.3% SFA intake, over 4-10yrs of follow-up in which 5,249 coronary events and 2,155 coronary deaths occurred. Replacing 5% of energy from SFA with PUFA was associated with a 13% relative risk reduction for coronary events [HR 0.87, 95% CI 0.77-0.97] reduced risk for coronary deaths by 26% [HR 0.74, 95% CI 0.61-0.89].
In a meta-analysis by Farvid et al. including 12 cohorts in the US, Europe, and Israel, comparing high vs. low intakes [median 6.4% to 1.5%, respectively] of LA was associated with a 15% [HR 0.85, 95% CI 0.78-0.92] relative reduction in risk for CHD events, and 21% [HR 0.79, 95% CI 0.71–0.89] reduction in risk for CHD death. In dose-response analysis the relationship between increasing LA as a percentage of energy and lower risk of CHD events and deaths was linear: each 5% increase in LA was associated with a 10% [RR 0.90, 95% CI 0.85–0.94] lower risk for events and 13% [RR 0.87, 95% CI 0.81–0.93] lower risk for CHD mortality.
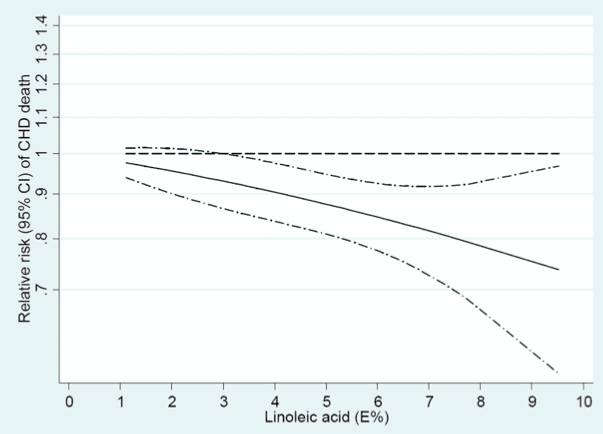
Figure from Farvid et al. illustrating linear association between increasing linoleic acid (as a percentage of energy) and reduction in risk for coronary heart disease.
This linear association was observed again by a paper published this year by Li et al., a meta-analysis including 21 prospective cohorts, 811,069 participants, 50,786 CVD deaths, and follow-up median duration from 4.9yrs to 30yrs. PUFA intake ranged from 1.1% to 11.6%, and each 5% increment of energy intake from LA was associated with a 7% [RR 0.93, 95% CI 0.91-0.95] reduction in CVD risk.
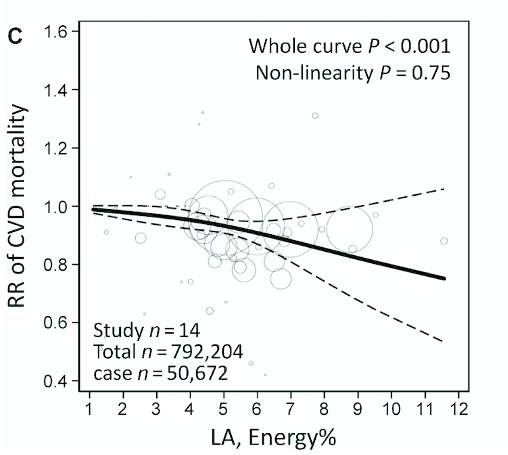
Figure from Li et al. illustrating linear association between increasing linoleic acid (as a percentage of energy) and reduction in cardiovascular disease mortality risk.
Finally, in a combined analysis of the Nurses’ Health Study and the Health Professionals Follow-Up Study, substitution of 5% energy from SFA with PUFA generally was associated with a 25% reduction in CHD mortality. Of particular note in this analysis was the hierarchy of effects of substation: if you contrast this with the figure from the Clarke et al. meta-analysis of 395 metabolic ward studies above, you’ll see that the effect of substitution mirrors that of the magnitude of effect of substitution on blood lipids. While there is no doubt more going on in the picture of cardio-metabolic health, this relationships provides biological plausibility to the reductions in CVD and CHD risk observed by increasing PUFA intake, particularly where it displaces dietary SFA.
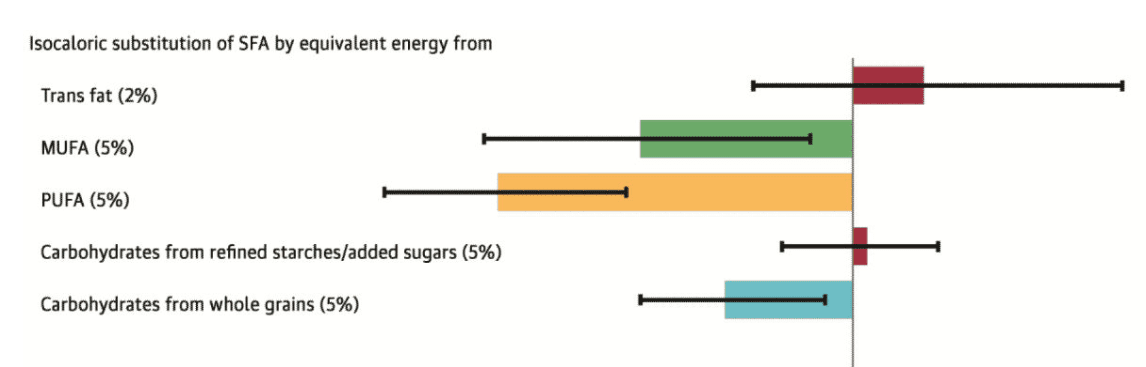
Figure from combined analysis of the NHS and HPFS by Li et al., illustrating the reduction in risk for coronary heart disease mortality associated with isocaloric replacement of saturated fat with other fat subtypes and carbohydrate subtypes. Replacement hierarchy evident as PUFA>MUFA>Complex Carbohydrate, consistent with data from metabolic ward studies of effects of replacement on blood lipids.
And what about dementia and Alzheimer’s Disease? In the Dutch Doetinchem Cohort Study, EPA and DHA were together associated with a 19% [OR 0.81, 95% CI 0.66-1.00] reduction in risk. These associations were not observed for other polyunsaturated fats, including the precursor to EPA and DHA, ALA, which suggests a particular role for EPA/DHA in the prevention of cognitive decline. This finding was repeated in the Chicago Health and Ageing Project [CHAP] study, which found that while neither ALA or EPA were associated with reduced risk, DHA specifically was associated with significant reductions in Alzheimer’s Disease risk [RR 0.30, 95% CI 0.10-0.90].
Cumulatively, the direction of effect in long-term prospective studies is consistent across populations in reducing risk for CVD and CHD outcomes, both events and deaths. This relationship has been shown to be linear for LA in separate analyses. In addition, the epidemiology on oily fish consumption and the long-chain omega-3 fatty acids, EPA and DHA, is one of the strongest associations for protective effects against dementia and Alzheimer’s Disease. Thus, from this body of prospective data we can state that the relationship is temporal, strong, consistently significant in the direction of reduced risk, and exhibits a dose-response. All of these factors support a benefit derived from dietary PUFA. Now let’s proceed to tissue biomarker studies.
Tissue Biomarker Studies:
Biomarker* studies are primarily prospective cohort studies, but are highly valuable pieces of the evidential puzzle.
In the Fatty Acid and Outcome Research Consortium [FORCE] of studies comprising 30 cohorts in which 76,356 fatty acid measurements of circulating or adipose tissue fatty acids from 68,659 participants across 13 countries, pooled analysis demonstrated that the highest levels of tissue LA were associated with a 23% [HR 0.77, 95% CI 0.69-0.86] reduction in risk for total CVD. Plasma levels of the dreaded arachidonic acid were also associated with a significant 21% [HR 0.79, 95% CI 0.67-0.93] reduction in risk for total CVD. But this had been previously demonstrated, in a meta-analysis of 10 studies on biomarkers of AA levels, circulating levels were associated with a 17% relative reduction in CVD risk [RR 0.83, 95% CI 0.74–0.92].
In the US Cardiovascular Health Study, in participants followed-up over 34,291 person-years of follow-up in which 1,994 deaths occurred, higher plasma phospholipid LA levels were associated with a significant 13% [HR 0.87, 95% CI 0.74–1.02] lower risk of all-cause mortality. In a Finnish cohort study, higher serum levels of LA were associated with a significant 46% [HR 0.54, 95% CI 0.40-0.74] relative risk reduction comparing the highest to lowest circulating biomarker levels. In the recent meta-analysis by Li et al., data on biomarkers of LA and mortality outcomes was available from 28 prospective cohorts, including 65,411 participants. In the analysis of all tissue compartments combined, increased tissue LA concentrations in pooled analysis was associated with a 11% [HR0.89, 95% CI 0.85-0.94] reduction in risk for CVD.
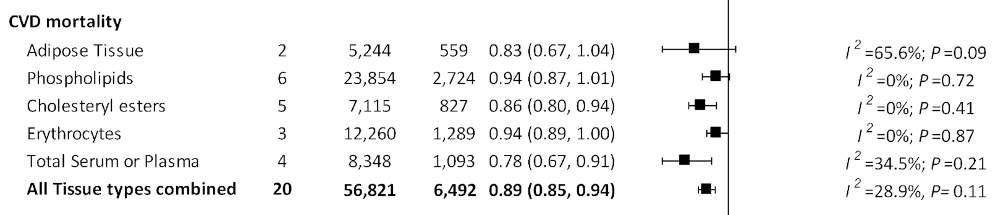
Forest plot from Li et al. demonstrating overall effect size associated with linoleic acid measures in various tissue compartments, and overall effect size.
Similarly, biomarker studies have provided mechanistic corroboration of prospective cohort findings in relation to dementia/AD. In the Framingham Offspring Study, participants with the lowest red blood cell DHA levels displayed significantly lower total cerebral brain volume and performed worse on cognitive testing, compared to the highest DHA levels. In the Women’s Health Initiative Brain MRI Study, women had MRI scans conducted 8yrs after baseline RBC omega-3 samples were collected. High baseline Omega-3 Index [combined DHA+EPA] levels correlated with a 2.1cm larger brain volume determined by MRI. This is, however, not to suggest that there is no role for EPA in protection against cognitive decline. EPA may be low in brain tissue itself, however, higher plasma levels of EPA have been associated with a 31% [HR 0.69, 95% CI 0.48-0.98] reduced depression-associated dementia risk, and higher cerebral plasma EPA levels associated with prevention of brain grey matter atrophy in the amygdala.
By conducting analyses of biomarkers of PUFA intake, i.e, objective in vivo measures of fatty acids, these studies add a level of certainty from epidemiology, where an RCT with thousands of participants and objective measures of diet would be infeasible. Nevertheless, there are people who will dismiss all of the foregoing in favour of rats and Sydney-Diet Heart, so let’s wrap this up with evidence from the hallowed sanctity that is the randomised controlled intervention trial.
Intervention Studies:
This is where the data comes full circle and appears unequivocal. In 2010, Mozaffarian et al. conducted a meta-analysis of controlled feeding studies with a median of 4.25yrs duration. In the overall pooled analysis of eight RCTs, there was a 19% [RR 0.81, 95% CI 0.70-0.95] relative reduction in risk for CHD events. Subgroup analysis by trial duration indicated that in interventions >4.25yrs there was a 27% [RR 0.73, 95% CI 0.61–0.87] reduction in CHD event risk. In substitution analysis, a mean increase of 10% PUFA in the intervention groups indicated that each 5% energy from SFA replaced with PUFA reduced LDL-C by an average of 0.25mmol/L [10mg/dL], and CVD risk decreased by 10% [RR 0.90, 95% CI 0.83–0.97]. The mean cholesterol reduction in the intervention arm compared to the control arms was was 0.76mmol/L [29 mg/dl], corresponding to a 24% [RR 0.76, 95% CI 0.62–0.93] reduction in CHD event risk for each 1mmol/L [38.6mg/dL] reduction in total cholesterol levels. Of particular note in this comprehensive meta-analysis was the correlation between the relative risk reduction for CHD from predicted changes in blood cholesterol from intervention studies, the reduction in CHD events in intervention studies, and the reduction in CHD risk observed in data from the Jakobsen et al. pooled analysis cited above.
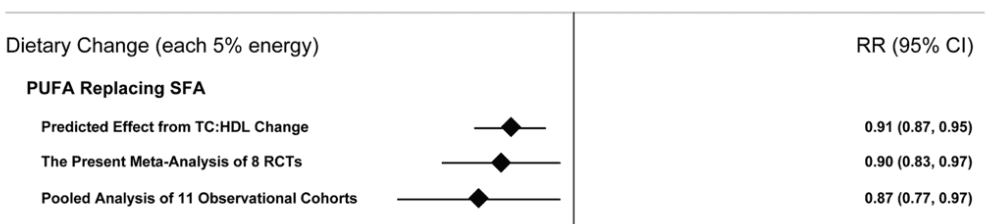
Figure from Mozaffarian et al. illustrating relative risk of coronary heart disease events from PUFA replacing SFA, including predicted effects on blood cholesterol from short-term interventions, the meta-analysis of interventions, and the data extracted from the pooled analysis by Jakobsen et al.
And we can look at the Hooper et al. updated meta-analysis of intervention studies from this year, which found that the replacement of SFA with PUFA was associated with a 27% [RR 0.73, 95% CI 0.58-0.92] reduction in risk for CVD events. We could look at the REDUCE-IT [Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial], which found that a high-dose EPA supplement given to patients in secondary prevention [i.e., they had already suffered a cardiovascular event] led to a 25% reduction in risk for a further cardiovascular event. We could look at the recent Hu et al. meta-analysis of omega-3 supplemental interventions in secondary prevention, which included REDUCE-IT, and found an 8% [HR 0.92, 95% CI 0.86-0.98] reduction in risk for CHD death and 12%[HR 0.88, 95% CI 0.83-0.94] reduction in risk for myocardial infarction. In dose-response meta-analysis, each 1g/d EPA/DHA resulted in a 17% [HR 0.92, 95% CI 0.88-0.97] reduced risk of CVD death.
The practical implication is that, in my opinion, anyone attempting to support an argument that replacing SFA with PUFA increases heart disease risk either hasn’t done their due diligence or is being wilfully ignorant of the totality of evidence because studies like Sydney Diet-Heart confirm their preconceived bias.
Conclusions
In order to sustain the case that PUFA intake is deleterious to human health, anyone advancing the argument has to provide a rebuttal to the multiple lines of evidence from RCTs, prospective cohort studies, and biomarker studies, which all converge to the same conclusion. They can’t.
In order to sustain such a case, the putative mechanisms of lipid peroxidation and inflammation have to be shown to relate to adverse health outcomes with supporting evidence of sufficient quality in humans. It isn’t there.
And in order to sustain such a case, the handful of studies that are cited in support of such an adverse effect in humans need to stand up to scrutiny of their methodology. They don’t.
A total body of evidence: ecological studies, prospective cohort studies, tissue biomarker studies, metabolic ward studies, intervention studies, and meta-analyse respectively of cohort studies, biomarker studies, metabolic ward studies, and intervention studies. Every line of evidence converges to a conclusion in favour of the role of polyunsaturated fats, including the omega-6 linoleic acid, and health outcomes in humans."
Thoughts on this article?
---------------------------------------------------------------------------------------------------------------------------------------
In addition to that:
The Five Things to Look for When Choosing a Fish Oil Supplement — Ben Greenfield
Ben Greenfield gives his list of five things to look for when choosing a fish oil supplement in this 2-minute audio clip
In short summary:
So something like WHC fish oil would suffice.
- Low in mercury
- It's in the triglyceride form (NOT ethyl ester form)
- It comes from small, cold water fish (preferably anchovies)
- It's preserved with astaxanthin or rosemary
- It's high in omega-3s
If a high-quality fish oil supplement contains vitamin E and/or rosemary as preservatives along with the EFA in the softgel and follows the other requirements, do you still doubt that it would be beneficial?
Capt Nirvana
Member
- Joined
- Mar 25, 2018
- Messages
- 108
I've written 12 e-books to counteract all of the above. Yellow Fat Disease (progressive lipofuscinosis) is an indicator of aging and almost all age-related diseases in man, other animals, insects, fish, shrimp, etc. Fortunately for full-spectrum omnivores like man, cumulative lipofuscinosis is an indicator of BIOLOGICAL aging, not CHRONOLOGICAL aging, as most mainstream authorities claim. (By the way, your puny list is the snowflake on the tip of the iceberg; there are over 40,000 mainstream studies on over 4 million people promoting your incorrect information.)
Is this Atom Bergstrom?I've written 12 e-books to counteract all of the above. Yellow Fat Disease (progressive lipofuscinosis) is an indicator of aging and almost all age-related diseases in man, other animals, insects, fish, shrimp, etc. Fortunately for full-spectrum omnivores like man, cumulative lipofuscinosis is an indicator of BIOLOGICAL aging, not CHRONOLOGICAL aging, as most mainstream authorities claim. (By the way, your puny list is the snowflake on the tip of the iceberg; there are over 40,000 mainstream studies on over 4 million people promoting your incorrect information.
Can you try it again and let me know if it works now? Just make sure you sign out and sign in before doing it.I don't know why I can't quote your posts but the message you posted above is bull****, Daniel.
Apologies for anyone running into this issue. Working on getting it squashed.
this kind of masturbation is weird to me. Why not just look at the available randomized controlled trials in humans and see directly that replacing saturated fat with seed oils is very deadly, to a degree one of the trials had to be stopped and another one modified mid-trial completely fraudulently? Masterjohn has a video about this if you want sources. I wonder why such trials are not done any longer? 
The deceptive aspect is in the mechanism of PUFA toxicity, it starts to show severely increased mortality somewhere approaching year 5 and get really bad at 7. It is hard to make a proper trial this long in today's age of attention deficit and corruption.
The deceptive aspect is in the mechanism of PUFA toxicity, it starts to show severely increased mortality somewhere approaching year 5 and get really bad at 7. It is hard to make a proper trial this long in today's age of attention deficit and corruption.
EMF Mitigation - Flush Niacin - Big 5 Minerals
Similar threads
- Replies
- 0
- Views
- 2K
- Replies
- 143
- Views
- 25K
- Replies
- 0
- Views
- 2K
- Replies
- 60
- Views
- 10K
- Replies
- 22
- Views
- 8K
- Replies
- 41
- Views
- 11K
- Replies
- 2
- Views
- 2K
- Replies
- 3
- Views
- 2K
- Replies
- 2
- Views
- 4K
- Replies
- 1
- Views
- 2K
- Replies
- 15
- Views
- 3K