SFAs play the leading role in 1 of the greatest controversies in nutrition science. Relative to PUFAs, SFAs generally increase circulating concentrations of LDL cholesterol, a risk factor for atherosclerotic cardiovascular disease (ASCVD). However, the purpose of regulatory mechanisms that control the diet-induced lipoprotein cholesterol dynamics is rarely discussed in the context of human adaptive biology. We argue that better mechanistic explanations can help resolve lingering controversies, with the potential to redefine aspects of research, clinical practice, dietary advice, public health management, and food policy. In this paper we propose a novel model, the homeoviscous adaptation to dietary lipids (HADL) model, which explains changes in lipoprotein cholesterol as adaptive homeostatic adjustments that serve to maintain cell membrane fluidity and hence optimal cell function. Due to the highly variable intake of fatty acids in humans and other omnivore species, we propose that circulating lipoproteins serve as a buffer to enable the rapid redistribution of cholesterol molecules between specific cells and tissues that is necessary with changes in dietary fatty acid supply. Hence, circulating levels of LDL cholesterol may change for nonpathological reasons. Accordingly, an SFA-induced raise in LDL cholesterol in healthy individuals could represent a normal rather than a pathologic response. These regulatory mechanisms may become disrupted secondarily to pathogenic processes in association with insulin resistance and the presence of other ASCVD risk factors, as supported by evidence showing diverging lipoprotein responses in healthy individuals as opposed to those with metabolic disorders such as insulin resistance and obesity. Corresponding with the model, we suggest alternative contributing factors to the association between elevated LDL cholesterol concentrations and ASCVD, involving dietary factors beyond SFAs, such as an increased endotoxin load from diet-gut microbiome interactions and subsequent chronic low-grade inflammation that interferes with fine-tuned signaling pathways.
 academic.oup.com
academic.oup.com
Comments
Limitations in the “homeoviscous adaptation to dietary lipids” model
Amar Laila
I read with interest the article by Zinöcker et al., in which the authors proposed a model to explain the observed pathologic increase in LDL-cholesterol concentrations in response to higher SFA intake. The model de-emphasized the role of SFAs and suggested that dysregulation of cholesterol uptake is caused by endotoxemia, which causes inflammation and, therefore, prevents cholesterol uptake via LDL-cholesterol receptors. The authors’ conclusion was that, if the model is verified, recommendations should focus on reducing intake of refined carbohydrates, which increases the growth of LPS-containing gut bacteria, but not reducing intake of SFAs.

The homeoviscous adaptation to dietary lipids (HADL) hypothesis is probably incorrect
Jacob J Christensen
Zinöcker, Svendsen, and Dankel recently presented their homeoviscous adaptation to dietary lipids (HADL) model (1). Briefly, their model can be summarized as follows: high dietary SFA and low PUFA intake causes changes to plasma membrane fluidity that drive transfer of free cholesterol frommembranes to lipoproteins, whereas a low dietary SFA and high PUFA intake does the opposite. The authors suggest several immediate implications for how we now should understand biology and the connection between diet and health.
In the following, we address issues and topics which suggest that the presented hypothesis is incorrect.
First, let us discuss the key questions posed by the authors, concerning the origin and disposal of LDL cholesterol, as this is established scientific knowledge (2). Cholesterol in plasma is transported in lipoproteins, and in humans, the majority is found in apoB-100-containing LDL particles. All apoB-100-containing lipoprotein particles originate from VLDLs, which are being continuously secreted by the liver. The main function of VLDL particles is to transport lipids, mainly triglycerides (energy), from the liver to peripheral tissues. LDL particles are the smallest VLDL remnants from which most of the triglycerides have been depleted via the function of lipoprotein lipase and hepatic lipase. Furthermore, LDL particles are continuously removed from circulation by hepatocytes via the LDL-receptor (LDLR)-dependent pathway; LDL particles can therefore accumulate in the plasma because of increased VLDL secretion and residence time or reduced LDL clearance. After degradation in lysosomes, LDL particle contents will be dispersed into various cellular pools. Cholesterol, for example, will be deposited as lipid droplets, incorporated into membranes, used for VLDL particle biosynthesis, or secreted into the bile ducts either as cholesterol or as bile salts to reach the gut for reabsorption or ultimate excretion (2).
Second, we would like to comment on a few of the statements about atherosclerosis. Although endothelial injury (“response-to-injury”) may contribute, the real driver of atherosclerotic progression is the subendothelial retention of apoB-100-containing lipoprotein particles (“response-to-retention”) which then launches a local, sterile inflammation (3). This means that inflammation is a consequence rather than a cause of lipid accumulation in the arterial wall. In contrast, the cholesterol molecules present in the circulation are not relevant to atherosclerosis per se. Furthermore, all apoB-100-containing particles are potentially atherogenic, and the degree of atherosclerosis progression is driven mainly by the cumulative exposure to atherogenic lipoproteins, which is determined by the absolute plasma concentration and the duration of exposure (“cholesterol burden”). Indeed, a persistently elevated concentration of LDL particles in plasma is harmful regardless of its cause (unfavorable genetic variants or an unhealthy diet), whereas a persistently low concentration is beneficial regardless of its cause [favorable genetic variants, a healthy diet, or pharmacotherapy using statins, proprotein convertase subtilisin-kexin 9 (PCSK9) inhibitors, or ezetimibe] (3).
Third, the authors should have considered the following topics while developing their hypothesis:
From basic science, animal models, genetic studies, and intervention trials, we know the most important determinants of the variability in plasma LDL cholesterol (3, 4). For example, number and activity of hepatic LDLRs (relevant genes include LDLR, PCSK9, APOE), LDL binding to LDLRs (LDLR, APOB), cholesterol biosynthesis [3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR)], hepatic bile acid synthesis [cholesterol 7 alpha-hydroxylase (CYP7A1)], and enterocyte uptake and secretion of cholesterol [Niemann-Pick C1-Like 1, ATP-binding cassette sub-family G member 5 and 8 (NPC1L1, ABCG5/G8)].
All human cells can produce the cholesterol they need, and cellular cholesterol deficiency occurs only when its synthesis is defective (5–7). For example, heterozygous and homozygous familial hypercholesterolemia subjects exhibit 50% and 100% reduced cellular LDL uptake, respectively, but show no symptoms consistent with cholesterol deficiency. In contrast, cholesterol synthesis defects such as Smith–Lemli–Opitz syndrome are detrimental already in fetal life.
In contrast to glucose homeostasis, no hormonal system has evolved to keep plasma cholesterol concentration within a narrow range. This is because every cell can synthesize cholesterol and, accordingly, no cell has an absolute requirement for cholesterol uptake (6–8). Therefore, plasma LDL-cholesterol concentrations can be extremely low (although not 0) with few adverse effects.
Because free cholesterol is toxic and human cells cannot degrade cholesterol molecules, there is a balance between whole-body cholesterol input through diet and biosynthesis, and output through utilization and excretion (2, 8).
Finally, the HADL hypothesis provides at least two testable implications. The first relates to dose-response. If the physicochemical properties of dietary fatty acids affect membrane fluidity, which then determines cholesterol accretion in membranes, which then causes plasma LDL cholesterol to increase or decrease, we should be able to predict the change in LDL cholesterol based on the physicochemical properties of dietary fatty acids. Theoretically, an incrementally higher melting point of dietary fatty acids would cause incremental increases in plasma LDL cholesterol, whereas an incrementally lower melting point would do the opposite. But from controlled interventions, we find no such relation. Among the SFAs, myristic acid (14:0) increases plasma LDL cholesterol most strongly [+0.071 mM per energy percent (E%) increase in the diet], followed by palmitic acid (16:0, +0.047 mM/E%), then lauric acid (12:0, +0.01 mM/E%) (9). Among the PUFAs, both linoleic acid (18:2n–6) and α-linolenic acid (18:3n–3) reduce plasma LDL cholesterol by equal amounts (−0.017 mM/E%) (9). And importantly, of the long-chain omega-3 fatty acids, neither EPA (20:5n–3) nor DHA (22:6n–3) affects plasma LDL cholesterol (4).
Another testable implication concerns the temporal sequence, an essential part of any cause–effect analysis. If dietary fatty acids affect membrane fluidity, which then determines cholesterol accretion in membranes, and which then causes plasma LDL cholesterol to increase or decrease, we should observe membrane remodeling before the changes in plasma LDL cholesterol. However, that is not what we observe in controlled interventions: whereas plasma LDL cholesterol responds rapidly to dietary change (days to weeks), changes in membrane lipid composition occur slowly (weeks to months) and depend largely on the rate of cell turnover (10).
In summary, the authors have presented a biased scientific rationale, and they have omitted information that directly contradicts their hypothesis. We find that the HADL hypothesis conflicts with established knowledge and, therefore, based on all the aforementioned considerations, we propose that the presented hypothesis is incorrect.
 academic.oup.com
academic.oup.com
Reply to JJ Christensen
Christensen et al. suggest that the hypothesis underlying the homeoviscous adaptation to dietary lipids (HADL) model (1) is incorrect. However, none of their arguments falsify the model, and recent research into the effects of dietary fatty acids on membrane remodeling, including cholesterol incorporation, supports it.
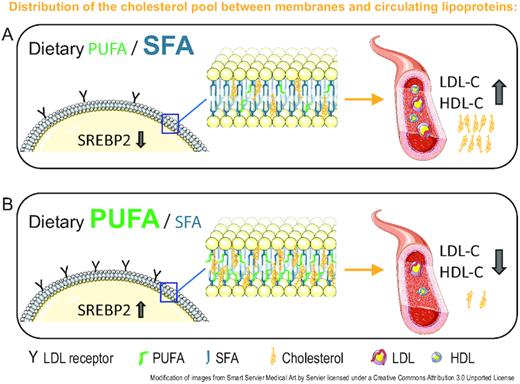
In the fields of biophysics and molecular biology, there is currently intensive research into the mechanisms that regulate plasma membrane fluidity and thereby cell function. Intriguingly, recent studies have revealed novel molecular sensors in specific membrane compartments that enable homeostatic membrane adaptations. These mechanisms involve complementary transcriptional regulators including sterol regulatory element-binding protein (SREBP) and are influenced by fatty acid supply (2, 3). However, despite the long-known essentiality of optimal plasma membrane function, the nature and purpose of regulatory mechanisms that remodel membrane lipids, in part determined by dietary fatty acid composition, remain insufficiently described. From the cell-centric perspectives of HADL, important new questions and knowledge gaps become evident and require further investigation.
We agree that the temporal (cause–effect) issue is important. But to properly understand the dynamics of cholesterol distribution between tissues and circulating lipoproteins, we propose an expanded view from single cell types—such as nonnucleated erythrocytes which are typically used as markers of longer-term fatty acid intake—to the homeostatic dynamics of fatty acid and cholesterol redistribution across different tissues/cell types.
Support for both rapid and tissue-specific fatty acid incorporation comes from a study on fish oil supplementation in rats, showing a doubling of DHA (22:6n–3) concentrations in myocardial cells already within 2 d, whereas erythrocytes showed considerably slower and more variable fatty acid incorporation (4). Furthermore, Levental et al. (5) recently demonstrated a surprisingly rapid lipid remodeling of mammalian cellular membranes after addition of exogenous DHA. Incorporation of DHA induced lipidome-wide remodeling, most notably upregulation of saturated membrane lipids and cholesterol, resulting in recovery of membrane packing and permeability. The cholesterol incorporation was concomitant with the acyl chain remodeling, seen already 1 h after DHA introduction and with peak levels at 4 h. These findings were also confirmed in vivo within a timeframe of 2 wk (although not measured earlier) (5). As stated by Levental et al. (5): “cholesterol is rarely considered alongside fatty acids and is a major component of the remodeling we report.” In sum, these empirical data demonstrate that changes in cell membrane lipids after altered fatty acid supply occur rapidly and are likely to help explain LDL-cholesterol changes observed in intervention studies. With the HADL model we propose that such adaptive changes at the cellular level are coupled with cholesterol uptake from and efflux to lipoproteins. These findings and concepts strongly motivate more research into the mechanisms that regulate plasma membrane lipid dynamics in response to diet, including temporal and cross-tissue relations.
Regarding the suggestion that the melting point of dietary fatty acids would predict the changes in cholesterol content of circulating lipoproteins, we must consider that:
fatty acids are incorporated into the membrane in a highly regulated manner (6);
modification of dietary fatty acids takes place before incorporation into cell membranes (7); and
dietary SFA may be incorporated into cell membranes to a lesser degree than dietary PUFA (SFA being more derived from endogenous synthesis) (5).
Hence, the effect on membrane fluidity and subsequent change in LDL-cholesterol concentrations cannot solely be extrapolated from the melting point of dietary fatty acids.
Regarding other proposed arguments by Christensen et al. against the plausibility of the HADL model, we have the following comments:
The authors list a number of determinants of variability of plasma LDL cholesterol. We would like to add that the expression and function of the proteins mentioned are highly influenced by diet, and hence must be evaluated according to dietary context.
The authors claim that all cells synthesize the cholesterol they need. This is incorrect, because only nucleated cells can synthesize cholesterol, resulting in different mechanisms of lipid incorporation into erythrocyte membranes. They also argue that the lack of classical cholesterol deficiency syndrome in familial hypercholesterolemia speaks for sufficient cholesterol synthesis to serve membrane needs. This argument ignores that cholesterol demand can increase with changing dietary conditions as shown by Levental et al. (5), and that select tissues might depend on other sources than endogenous synthesis for sufficient cholesterol supply, as demonstrated by the role of scavenger receptor class B type I (SR-BI) in the endothelium of mice (8). The transcription and expression of proteins central to uptake and synthesis of cholesterol, such as the LDL receptor, SR-BI, and β-hydroxy-β-methylglutaryl coenzyme A (HMG-CoA) reductase, are highly variable between cells and tissues and indicate that different cell types have evolved distinct strategies for cholesterol supply.
The authors moreover interpret the lack of hormonal regulation of plasma cholesterol concentrations as evidence for sufficient synthesis in all tissues. The lack of such a direct regulation may just as well indicate that elevation in plasma cholesterol concentrations is not detrimental per se, so it is not a valid argument. In any case, in the presence of key risk factors associated with modern diets, such as insulin resistance and inflammation, functional cholesterol homeostasis can be disturbed (9).
Christensen et al. claim that LDL-cholesterol concentrations can be extremely low with few adverse effects. Although this is observed in genetic studies and clinical drug trials, there is also documentation that individuals with low serum cholesterol concentrations show greater risk of cancer and respiratory and gastrointestinal diseases (10), supporting the importance of considering cholesterol metabolism in a whole-body perspective.
Other points raised by Christensen et al. are not necessarily in conflict with (or relevant in light of) the HADL model. Although we appreciate that some of the inferences on implications hinge on the extent to which HADL determines circulating LDL cholesterol, and although LDL cholesterol is a risk factor for atherosclerotic cardiovascular disease (ASCVD), it is relevant to discuss other aspects of ASCVD pathogenesis congruent with a cell-centric, homeostatic view of diet-dependent cholesterol dynamics.
Taken together, experimental data support the HADL model and encourage further testing. Although all aspects of the HADL model have not yet been experimentally tested, the promising concepts and clear knowledge gaps emerging from our model call for more experimental evidence (such as the recent findings mentioned above), and should not be dismissed merely by theoretical arguments.

The homeoviscous adaptation to dietary lipids (HADL) model explains controversies over saturated fat, cholesterol, and cardiovascular disease risk
ABSTRACT. SFAs play the leading role in 1 of the greatest controversies in nutrition science. Relative to PUFAs, SFAs generally increase circulating concentrati
academic.oup.com
Comments
Limitations in the “homeoviscous adaptation to dietary lipids” model
Amar Laila
I read with interest the article by Zinöcker et al., in which the authors proposed a model to explain the observed pathologic increase in LDL-cholesterol concentrations in response to higher SFA intake. The model de-emphasized the role of SFAs and suggested that dysregulation of cholesterol uptake is caused by endotoxemia, which causes inflammation and, therefore, prevents cholesterol uptake via LDL-cholesterol receptors. The authors’ conclusion was that, if the model is verified, recommendations should focus on reducing intake of refined carbohydrates, which increases the growth of LPS-containing gut bacteria, but not reducing intake of SFAs.
The homeoviscous adaptation to dietary lipids (HADL) hypothesis is probably incorrect
Jacob J Christensen
Zinöcker, Svendsen, and Dankel recently presented their homeoviscous adaptation to dietary lipids (HADL) model (1). Briefly, their model can be summarized as follows: high dietary SFA and low PUFA intake causes changes to plasma membrane fluidity that drive transfer of free cholesterol frommembranes to lipoproteins, whereas a low dietary SFA and high PUFA intake does the opposite. The authors suggest several immediate implications for how we now should understand biology and the connection between diet and health.
In the following, we address issues and topics which suggest that the presented hypothesis is incorrect.
First, let us discuss the key questions posed by the authors, concerning the origin and disposal of LDL cholesterol, as this is established scientific knowledge (2). Cholesterol in plasma is transported in lipoproteins, and in humans, the majority is found in apoB-100-containing LDL particles. All apoB-100-containing lipoprotein particles originate from VLDLs, which are being continuously secreted by the liver. The main function of VLDL particles is to transport lipids, mainly triglycerides (energy), from the liver to peripheral tissues. LDL particles are the smallest VLDL remnants from which most of the triglycerides have been depleted via the function of lipoprotein lipase and hepatic lipase. Furthermore, LDL particles are continuously removed from circulation by hepatocytes via the LDL-receptor (LDLR)-dependent pathway; LDL particles can therefore accumulate in the plasma because of increased VLDL secretion and residence time or reduced LDL clearance. After degradation in lysosomes, LDL particle contents will be dispersed into various cellular pools. Cholesterol, for example, will be deposited as lipid droplets, incorporated into membranes, used for VLDL particle biosynthesis, or secreted into the bile ducts either as cholesterol or as bile salts to reach the gut for reabsorption or ultimate excretion (2).
Second, we would like to comment on a few of the statements about atherosclerosis. Although endothelial injury (“response-to-injury”) may contribute, the real driver of atherosclerotic progression is the subendothelial retention of apoB-100-containing lipoprotein particles (“response-to-retention”) which then launches a local, sterile inflammation (3). This means that inflammation is a consequence rather than a cause of lipid accumulation in the arterial wall. In contrast, the cholesterol molecules present in the circulation are not relevant to atherosclerosis per se. Furthermore, all apoB-100-containing particles are potentially atherogenic, and the degree of atherosclerosis progression is driven mainly by the cumulative exposure to atherogenic lipoproteins, which is determined by the absolute plasma concentration and the duration of exposure (“cholesterol burden”). Indeed, a persistently elevated concentration of LDL particles in plasma is harmful regardless of its cause (unfavorable genetic variants or an unhealthy diet), whereas a persistently low concentration is beneficial regardless of its cause [favorable genetic variants, a healthy diet, or pharmacotherapy using statins, proprotein convertase subtilisin-kexin 9 (PCSK9) inhibitors, or ezetimibe] (3).
Third, the authors should have considered the following topics while developing their hypothesis:
From basic science, animal models, genetic studies, and intervention trials, we know the most important determinants of the variability in plasma LDL cholesterol (3, 4). For example, number and activity of hepatic LDLRs (relevant genes include LDLR, PCSK9, APOE), LDL binding to LDLRs (LDLR, APOB), cholesterol biosynthesis [3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR)], hepatic bile acid synthesis [cholesterol 7 alpha-hydroxylase (CYP7A1)], and enterocyte uptake and secretion of cholesterol [Niemann-Pick C1-Like 1, ATP-binding cassette sub-family G member 5 and 8 (NPC1L1, ABCG5/G8)].
All human cells can produce the cholesterol they need, and cellular cholesterol deficiency occurs only when its synthesis is defective (5–7). For example, heterozygous and homozygous familial hypercholesterolemia subjects exhibit 50% and 100% reduced cellular LDL uptake, respectively, but show no symptoms consistent with cholesterol deficiency. In contrast, cholesterol synthesis defects such as Smith–Lemli–Opitz syndrome are detrimental already in fetal life.
In contrast to glucose homeostasis, no hormonal system has evolved to keep plasma cholesterol concentration within a narrow range. This is because every cell can synthesize cholesterol and, accordingly, no cell has an absolute requirement for cholesterol uptake (6–8). Therefore, plasma LDL-cholesterol concentrations can be extremely low (although not 0) with few adverse effects.
Because free cholesterol is toxic and human cells cannot degrade cholesterol molecules, there is a balance between whole-body cholesterol input through diet and biosynthesis, and output through utilization and excretion (2, 8).
Finally, the HADL hypothesis provides at least two testable implications. The first relates to dose-response. If the physicochemical properties of dietary fatty acids affect membrane fluidity, which then determines cholesterol accretion in membranes, which then causes plasma LDL cholesterol to increase or decrease, we should be able to predict the change in LDL cholesterol based on the physicochemical properties of dietary fatty acids. Theoretically, an incrementally higher melting point of dietary fatty acids would cause incremental increases in plasma LDL cholesterol, whereas an incrementally lower melting point would do the opposite. But from controlled interventions, we find no such relation. Among the SFAs, myristic acid (14:0) increases plasma LDL cholesterol most strongly [+0.071 mM per energy percent (E%) increase in the diet], followed by palmitic acid (16:0, +0.047 mM/E%), then lauric acid (12:0, +0.01 mM/E%) (9). Among the PUFAs, both linoleic acid (18:2n–6) and α-linolenic acid (18:3n–3) reduce plasma LDL cholesterol by equal amounts (−0.017 mM/E%) (9). And importantly, of the long-chain omega-3 fatty acids, neither EPA (20:5n–3) nor DHA (22:6n–3) affects plasma LDL cholesterol (4).
Another testable implication concerns the temporal sequence, an essential part of any cause–effect analysis. If dietary fatty acids affect membrane fluidity, which then determines cholesterol accretion in membranes, and which then causes plasma LDL cholesterol to increase or decrease, we should observe membrane remodeling before the changes in plasma LDL cholesterol. However, that is not what we observe in controlled interventions: whereas plasma LDL cholesterol responds rapidly to dietary change (days to weeks), changes in membrane lipid composition occur slowly (weeks to months) and depend largely on the rate of cell turnover (10).
In summary, the authors have presented a biased scientific rationale, and they have omitted information that directly contradicts their hypothesis. We find that the HADL hypothesis conflicts with established knowledge and, therefore, based on all the aforementioned considerations, we propose that the presented hypothesis is incorrect.
The homeoviscous adaptation to dietary lipids (HADL) hypothesis is probably incorrect
Dear Editor:
academic.oup.com
Reply to JJ Christensen
Christensen et al. suggest that the hypothesis underlying the homeoviscous adaptation to dietary lipids (HADL) model (1) is incorrect. However, none of their arguments falsify the model, and recent research into the effects of dietary fatty acids on membrane remodeling, including cholesterol incorporation, supports it.
In the fields of biophysics and molecular biology, there is currently intensive research into the mechanisms that regulate plasma membrane fluidity and thereby cell function. Intriguingly, recent studies have revealed novel molecular sensors in specific membrane compartments that enable homeostatic membrane adaptations. These mechanisms involve complementary transcriptional regulators including sterol regulatory element-binding protein (SREBP) and are influenced by fatty acid supply (2, 3). However, despite the long-known essentiality of optimal plasma membrane function, the nature and purpose of regulatory mechanisms that remodel membrane lipids, in part determined by dietary fatty acid composition, remain insufficiently described. From the cell-centric perspectives of HADL, important new questions and knowledge gaps become evident and require further investigation.
We agree that the temporal (cause–effect) issue is important. But to properly understand the dynamics of cholesterol distribution between tissues and circulating lipoproteins, we propose an expanded view from single cell types—such as nonnucleated erythrocytes which are typically used as markers of longer-term fatty acid intake—to the homeostatic dynamics of fatty acid and cholesterol redistribution across different tissues/cell types.
Support for both rapid and tissue-specific fatty acid incorporation comes from a study on fish oil supplementation in rats, showing a doubling of DHA (22:6n–3) concentrations in myocardial cells already within 2 d, whereas erythrocytes showed considerably slower and more variable fatty acid incorporation (4). Furthermore, Levental et al. (5) recently demonstrated a surprisingly rapid lipid remodeling of mammalian cellular membranes after addition of exogenous DHA. Incorporation of DHA induced lipidome-wide remodeling, most notably upregulation of saturated membrane lipids and cholesterol, resulting in recovery of membrane packing and permeability. The cholesterol incorporation was concomitant with the acyl chain remodeling, seen already 1 h after DHA introduction and with peak levels at 4 h. These findings were also confirmed in vivo within a timeframe of 2 wk (although not measured earlier) (5). As stated by Levental et al. (5): “cholesterol is rarely considered alongside fatty acids and is a major component of the remodeling we report.” In sum, these empirical data demonstrate that changes in cell membrane lipids after altered fatty acid supply occur rapidly and are likely to help explain LDL-cholesterol changes observed in intervention studies. With the HADL model we propose that such adaptive changes at the cellular level are coupled with cholesterol uptake from and efflux to lipoproteins. These findings and concepts strongly motivate more research into the mechanisms that regulate plasma membrane lipid dynamics in response to diet, including temporal and cross-tissue relations.
Regarding the suggestion that the melting point of dietary fatty acids would predict the changes in cholesterol content of circulating lipoproteins, we must consider that:
fatty acids are incorporated into the membrane in a highly regulated manner (6);
modification of dietary fatty acids takes place before incorporation into cell membranes (7); and
dietary SFA may be incorporated into cell membranes to a lesser degree than dietary PUFA (SFA being more derived from endogenous synthesis) (5).
Hence, the effect on membrane fluidity and subsequent change in LDL-cholesterol concentrations cannot solely be extrapolated from the melting point of dietary fatty acids.
Regarding other proposed arguments by Christensen et al. against the plausibility of the HADL model, we have the following comments:
The authors list a number of determinants of variability of plasma LDL cholesterol. We would like to add that the expression and function of the proteins mentioned are highly influenced by diet, and hence must be evaluated according to dietary context.
The authors claim that all cells synthesize the cholesterol they need. This is incorrect, because only nucleated cells can synthesize cholesterol, resulting in different mechanisms of lipid incorporation into erythrocyte membranes. They also argue that the lack of classical cholesterol deficiency syndrome in familial hypercholesterolemia speaks for sufficient cholesterol synthesis to serve membrane needs. This argument ignores that cholesterol demand can increase with changing dietary conditions as shown by Levental et al. (5), and that select tissues might depend on other sources than endogenous synthesis for sufficient cholesterol supply, as demonstrated by the role of scavenger receptor class B type I (SR-BI) in the endothelium of mice (8). The transcription and expression of proteins central to uptake and synthesis of cholesterol, such as the LDL receptor, SR-BI, and β-hydroxy-β-methylglutaryl coenzyme A (HMG-CoA) reductase, are highly variable between cells and tissues and indicate that different cell types have evolved distinct strategies for cholesterol supply.
The authors moreover interpret the lack of hormonal regulation of plasma cholesterol concentrations as evidence for sufficient synthesis in all tissues. The lack of such a direct regulation may just as well indicate that elevation in plasma cholesterol concentrations is not detrimental per se, so it is not a valid argument. In any case, in the presence of key risk factors associated with modern diets, such as insulin resistance and inflammation, functional cholesterol homeostasis can be disturbed (9).
Christensen et al. claim that LDL-cholesterol concentrations can be extremely low with few adverse effects. Although this is observed in genetic studies and clinical drug trials, there is also documentation that individuals with low serum cholesterol concentrations show greater risk of cancer and respiratory and gastrointestinal diseases (10), supporting the importance of considering cholesterol metabolism in a whole-body perspective.
Other points raised by Christensen et al. are not necessarily in conflict with (or relevant in light of) the HADL model. Although we appreciate that some of the inferences on implications hinge on the extent to which HADL determines circulating LDL cholesterol, and although LDL cholesterol is a risk factor for atherosclerotic cardiovascular disease (ASCVD), it is relevant to discuss other aspects of ASCVD pathogenesis congruent with a cell-centric, homeostatic view of diet-dependent cholesterol dynamics.
Taken together, experimental data support the HADL model and encourage further testing. Although all aspects of the HADL model have not yet been experimentally tested, the promising concepts and clear knowledge gaps emerging from our model call for more experimental evidence (such as the recent findings mentioned above), and should not be dismissed merely by theoretical arguments.
:")