GorillaHead
Member
So lets Revise. Nlrp3 is activated. Either by physical trauma to cells. Endotoxin. Bacteria. Fungi. And ?
Follow along with the video below to see how to install our site as a web app on your home screen.
Note: This feature may not be available in some browsers.
Click Here if you want to upgrade your account
If you were able to post but cannot do so now, send an email to admin at raypeatforum dot com and include your username and we will fix that right up for you.
So lets Revise. Nlrp3 is activated. Either by physical trauma to cells. Endotoxin. Bacteria. Fungi. And ?
This did not answer my question at all. I just went further down the pathway of inflammation."Gross calcification generally follows the fibrosis that is produced by inflammation." Ray Peat
“All cell death is characterized by an increase of intracellular calcium….” “Increase of cytoplasmic free calcium may therefore be called ‘the final common path’ of cell disease and cell death. Aging as a background of diseases is also characterized by an increase of intracellular calcium. Diseases typically associated with aging include hypertension, arteriosclerosis, diabetes mellitus and dementia.” -Fujita, 1991
.............
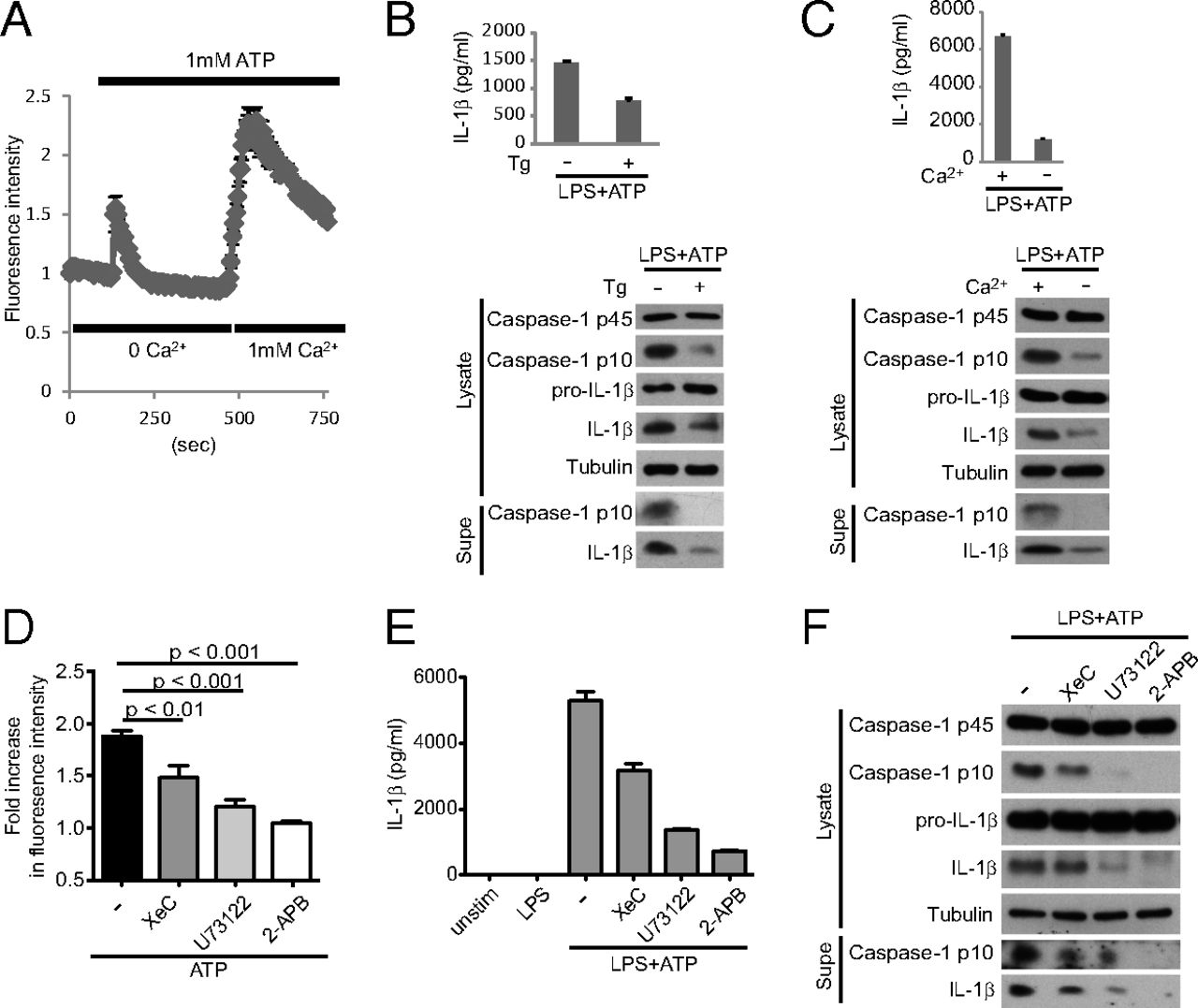
Critical role for calcium mobilization in activation of the NLRP3 inflammasome
Abstract
The NLRP3 (nucleotide-binding domain, leucine-rich-repeat-containing family, pyrin domain-containing 3) inflammasome mediates production of inflammatory mediators, such as IL-1β and IL-18, and as such is implicated in a variety of inflammatory processes, including infection, sepsis, autoinflammatory diseases, and metabolic diseases. The proximal steps in NLRP3 inflammasome activation are not well understood. Here we elucidate a critical role for Ca2+ mobilization in activation of the NLRP3 inflammasome by multiple stimuli. We demonstrate that blocking Ca2+ mobilization inhibits assembly and activation of the NLRP3 inflammasome complex, and that during ATP stimulation Ca2+ signaling is pivotal in promoting mitochondrial damage. C/EPB homologous protein, a transcription factor that can modulate Ca2+ release from the endoplasmic reticulum, amplifies NLRP3 inflammasome activation, thus linking endoplasmic reticulum stress to activation of the NLRP3 inflammasome. Our findings support a model for NLRP3 inflammasome activation by Ca2+-mediated mitochondrial damage.
Inflammation must be tightly regulated to meet the demands of host defense and stress adaptation while limiting immunopathology and the detrimental effects of chronic inflammation. Sensors of the innate immune system, such as pattern recognition receptors, couple detection of proinflammatory triggers to induction of inflammation in both adaptive and maladaptive settings, so the regulation of this interaction is absolutely critical in control of inflammation (1). A particularly intriguing “sensor” is the NLRP3 (nucleotide-binding domain, leucine-rich-repeat-containing family, pyrin domain-containing 3) inflammasome complex, which detects cellular stress in a variety of infectious and “sterile” settings and links this recognition to the induction of a critical inflammatory pathway (2⇓⇓–5).
The NLRP3 inflammasome is a cytoplasmic complex consisting of the regulatory subunit NLRP3, the adaptor ASC, and the effector subunit caspase-1. Stimulus dependent assembly of the complex activates the proteolytic activity of caspase-1, which is required for the processing and secretion of the inflammatory cytokines IL-1β and IL-18 (2⇓⇓–5). In recent years, many different molecules have been shown to activate the NLRP3 inflammasome complex. These molecules include: extracellular ATP released from dying cells (6); alum, the adjuvant activity in human vaccines (7); nigericin, a bacterial toxin with potassium ionophore activity (6); and cholesterol crystals (8), which contribute to the development of atherosclerotic plaques. Such findings underscore the critical role of the NLRP3 inflammasome in inducing inflammation in a variety of diseases.
How these diverse stimuli activate the NLRP3 inflammasome is not clear, but the prevailing view is that they generate some signal of cellular stress that is often referred to as signal 2. Although signal 1 is important for transcriptional up-regulation of the NLRP3 subunit and is typically provided by signaling through Toll-like receptors (9), three cellular processes have been suggested to provide signal 2 (2, 3, 10). The first is K+ efflux, which has been linked to most/all NLRP3 inflammasome activators, but just how K+ efflux can activate the inflammasome in a specific manner is not obvious; moreover, modulating the ionic milieu in other ways also affects NLRP3 inflammasome activation (11, 12). Second, crystals trigger phagolysosomal membrane damage/rupture (13); but how this is linked to the proximal steps in NLRP3 inflammasome activation is not clear, nor is its relevance for stimuli that are not phagocytosed. Finally, other studies implicate mitochondrial damage, including increased mitochondrial reactive oxygen species (mROS) production, loss of membrane potential (ΔΨ), and release of mtDNA into the cytosol (14⇓–16). Importantly, how NLRP3 inflammasome activators trigger mitochondrial damage has not been characterized.
Ca2+ mobilization controls diverse cellular processes, including proliferation and differentiation, transcription, cellular metabolism, and cell death (17). Such Ca2+ mobilization is made possible by maintaining low levels of cytoplasmic Ca2+ at basal state, so that signal-dependent influx of Ca2+ from the extracellular space and intracellular Ca2+ stores (e.g., endoplasmic reticulum, ER) leads to a rapid increase in cytoplasmic Ca2+ levels, activation of Ca2+ binding proteins, and induction of various cellular responses. A key player in Ca2+ signaling is the mitochondria, which take up Ca2+ from the ER or the extracellular space to regulate spatiotemporal patterns of Ca2+ signaling (18⇓–20). However, excessive or sustained mitochondrial Ca2+ uptake can lead to mitochondrial damage and cell death (21⇓–23).
In this article, we show that activation of the NLRP3 inflammasome requires Ca2+ signaling. Several NLRP3 inflammasome activators mobilize Ca2+, disruption of which inhibits NLRP3 inflammasome activation. During ATP stimulation, the crucial role of Ca2+ mobilization is in inducing mitochondrial damage. C/EPB homologous protein (CHOP), a protein known to regulate Ca2+ release from the ER during ER stress, amplifies NLRP3 inflammasome activation.
Critical role for calcium mobilization in activation of the NLRP3 inflammasome
The NLRP3 (nucleotide-binding domain, leucine-rich-repeat-containing family, pyrin domain-containing 3) inflammasome mediates production of inflammatory mediators, such as IL-1β and IL-18, and as such is implicated in a variety of inflammatory processes, including infection, sepsis...www.pnas.org
This did not answer my question at all. I just went further down the pathway of inflammation.
So far I haven't really seen any profound regrowth stories on this forum (eg returning to dense nw1).
I think Danny and Ray are correct, though I wonder if someone who has a better understanding of biochemistry than i do could possibly integrate this guys theory into our bioenergetic view:
(i imagine its the subtle piece of the puzzle that allows some of us to revert multiple norwoods. maybe not idrk lol)
here's a really interesting comment on a Danny Roddy video:
1:
Mark Panbecker:
"It only took me 17 years and and about 50K pubmed abstracts to find out the SAME mechanism that causes gout ALSO is the same mechanism that causes male pattern baldness. Castration prevents both gout and mpb. The mechanism or VERY close is: NLRP3 inflammasome activation which involves casp1 activation(which is higher in mpb scalp). Problem with too much casp1 is that it cleaves(chews up) the glucocorticoid receptor. Inflammation and autoimmunity are well known with NLRP3 activation. Without glucorticoid receptor function(I believe) you get AR upreg stepping in as a surrogate for a dysfunctional GC receptor. Glucocorticoid resistance is THEE major problem in hair loss. Autophagy prevents the NLRP3 cascade. That's why you see also babies sometimes born like a mpb shape even in girls because sometimes the mother has high GC levels in late term."
- I saw in a Peat facebook group a woman claiming her husband growing his hairline back after fasting
- casp1 in aga scalps study Caspase-1 level is higher in the scalp in androgenetic alopecia - PubMed
- this may explain some of the finasteride/ mtf transition successful regrowth
- its been mentioned on this forum that a finasteride user kept great hair but aged poorly in other ways
- could this mean there is a very specific type of inflammation cascade occuring in the scalps of balding men?
- some hypothyroid people maintain really great hair. Some people recover their stress/thyroid health to ideal / hyper yet struggle to gain significant regrowth
- in other words, could this make sense of how some unhealthy people can keep great hair?
2:
"Mark Panbecker4 years ago
Read this link about lyme disease(activates NLRP3) and male pattern baldness initiation. https://www.nczonline.net/blog/2014/04/02/i-have-lyme-disease/ Also immortal hair has some college guys suddenly develop mpb in their mold filled apartment. NLRP3 activated by microbes, cholesterol crystals, low PH, numerous things."
- a possible connection with Danny - genetically some have stress physiology that is susceptible to this "NLRP3 cascade"
- btw im not suggesting castration as a solution lmao, maybe there is some peaty method that should be doubled down on in particular to allow sufficient / youth like autophagy of "NLRP3 inflammasome" (?)
3:
"Mark Panbecker3 years ago
I never said GOUT was the cause of mpb. NLRP3 is the key pathway to look at for gout and mpb.Castration cures both. Autophagy (lack of it) it the real problem because dht downregulates it thru microRNA 221. Autophagy stops the nlrp3 from firing. Look at all drugs that cause hair growth just "happen" to increase autophagy."
possibly related forum thread?: Hair Regrowth After Glucocorticoids - Any Ideas Why?
dafaq?
Autophagy is essential for maintaining the growth of a human (mini-)organ: Evidence from scalp hair follicle organ culture
Autophagy is essential for maintaining the growth of a human (mini-)organ: Evidence from scalp hair follicle organ culture
link to the Danny Roddy video where you can find the discussion towards the bottom of the comment section:
(sorry if my writing is scattered)
surely we can solve MPB completely and for good. (Btw i like to imagine that if the Ray Peat / pro metabolic community can legitimately surpass the mainstream big 3 'treatments' - itd lead to a huge awakening in the masses view of mainstream pharmaceuticals/thyroid/scientism)
id love to hear what you folks think of this.
PirfenidoneThis true. And the RPF is not the best place to discuss hormesis since Peat is not too fond of the concept.
There is a lot of material available about autophagy, it’s role in aging/anti-aging and why and how it evolved and functions.
It’s the one point that I personally cannot square well with Peats focus on keeping a super energy metabolism. According to conventional knowledge, that would cause faster aging and disease - we have had many topics here why that also is not entirely true. But if we figure that out we are in for the novel prize 10 years in a row.
Anyway it seems more promising to use the autophagy mimicry by some substances to help the scalp on cellular level. Not amount of fasting or excervisd will bring back hair

 www.ncbi.nlm.nih.gov
www.ncbi.nlm.nih.gov
Have u tried the gel?Pirfenidone
This is the answer
It's even sold as a gel for scar removal.
This is how you stop all inflammation and reverse fibrosis.
Have u tried the gel?
Ive been using topical iodine. Too early to say anything. But it apparently regenerates scars. One guy on the internet reported some benefit on slick bald areasseems hard to find
it is sold as KitosCell
perhaps make your own by getting the powder?
Ive been using topical iodine. Too early to say anything. But it apparently regenerates scars. One guy on the internet reported some benefit on slick bald areas
True. Most natural stuff isnt strong enoughIt could work. But things like that are guesswork.
may as well for the most direct method - an approved pharmaceutical drug.
True. Most natural stuff isnt strong enough
Those are enzyme that breakdown things. They are honestly unlikely to inhibit processes in action.I was writing a few months ago about people using serrapeptase/bromelain, literally used in the burns unit in hospitals
But I still think this drug would be even better.
Those are enzyme that breakdown things. They are honestly unlikely to inhibit processes in action.
I ordered 10 grams will trial.It looks like KitosCell is available on Amazon in the US but it's pretty expensive.
How much did you pay? The 10 gram is $29.73 on Amazon right now which seems like a mistake because it's in the $200s everywhere else.I ordered 10 grams will trial.