Texon
Member
- Joined
- Nov 28, 2016
- Messages
- 671
@noordinary Haidut, FWIW I took one pill of this stuff and felt horrible. That was that as they say for me. And, I'm positive it wasn't the NR that made me feel that way. I am very interested to learn more about Cytoflavin though. Does anyone know of a reliable source for it?Yeah, unfortunately they are still pursuing the same old and discredited theory that activating sirtuins is the key to reverse aging and disease. Pterostilbene is just a more bioavailable form of resveratrol and Peat wrote a whole article on the resveratrol scam. I posted about it a few times too. If you search the forum for resveratrol you will find the threads. The infamous drug Vioxx, which killed quite a few people is also a stilbene and most/all stilbenes (including resveratrol) are estrogenic.
Aside from the nicotinamide riboside being crazy expensive for no good reason, a much better supplement to raise NAD/NADH ratio and achieve a host of other benefits would be a combination of plan niacinamide with methylene blue.
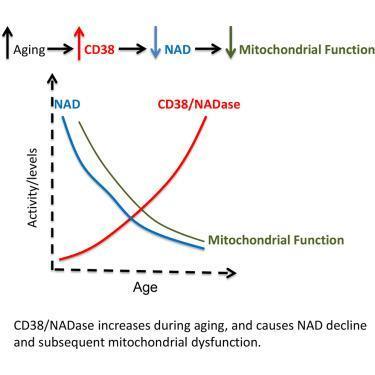
:") Hard to get better than that - increases NAD and decreases its degradation and consumption by all sort of enzymes like PARP for example.
Hard to get better than that - increases NAD and decreases its degradation and consumption by all sort of enzymes like PARP for example.