A gem. Fascinating read,
@haidut
 www.frontiersin.org
www.frontiersin.org
 Ruben Boon1,2,3*
Ruben Boon1,2,3*
Cellular metabolism is the cluster of enzymatic reactions that occur in cells to transform nutrients such as glucose, fatty acids, amino acids, and vitamins into cellular components, energy, and reducing power. As metabolites can freely diffuse through the relatively large nuclear pores, the nucleus has been proposed to be in metabolic homeostasis with the cytoplasm (Wente and Rout, 2010; Cambronne et al., 2016). Yet, fluctuations in cytoplasmic metabolites should not lead to random epigenetic changes in the nucleus. Furthermore, both the cytoplasm and nucleus represent highly crowded environments where molecular bindings and collisions severely limit the diffusion of molecules (Ellis, 2001; Richter et al., 2008). It is therefore unlikely that passive diffusion of metabolites from the cytoplasm into the nucleus forms the basis of the metabolism-epigenetic link.
In this review, I explore where the metabolic fuel of epigenetic modifiers might be produced. First, I show a correlation between epigenetic marks and cytoplasmic metabolism. Second, I review studies indicating a distinct nuclear metabolism based on the import of metabolic enzymes. Last, I elaborate on observations indicating further subnuclear compartmentalization based on protein complexation and phase separation.
Diet is therefore increasingly appreciated as an additional way to target the epigenome to influence inflammation (Wang et al., 2018), metabolic disease (Yuan et al., 2018), and neurogenesis defects (Benjamin et al., 2017).
Cellular metabolism is not only regulated by cell-intrinsic signaling pathways. Given that metabolic pathways consume nutrients, there is extensive cross-reactivity between cellular metabolism and the composition of the nutrient microenvironment (Elia and Haigis, 2021). For example, in the pyruvate-and proline-rich lung microenvironment, metastatic breast cancer cells preferentially catabolise these nutrients to sustain energy production and the production of extracellular matrix components (Elia et al., 2017; Elia et al., 2019). Additionally, lactate buildup in metabolically active and poorly vascularized non-small-cell lung cancers (NSCLCs), induces the use of this “waste” product as a substantial carbon source for fueling mitochondrial oxidative metabolism (Faubert et al., 2017). Last, nutrient depletion observed in most tumors leads to metabolic inactivation of immune cells. T cells are metabolically outcompeted by tumor cells in terms of glucose (Cascone et al., 2018), methionine (Bian et al., 2020), and arginine catabolism (Geiger et al., 2016).
The epigenome thus lies between the nutrient microenvironment and the regulation of cell fate. In this way, epigenetics might act as a metabolic antenna that probes cellular metabolism and translates environmental changes into transcriptional responses. Although the exact modifier was not identified, recently, a novel histone modification, histone lactylation, was indeed found to modulate wound healing and homeostasis of macrophages as a response to high levels of lactate as found in sepsis and hypoxia (Zhang et al., 2019). These findings also translate to the lactate-high tumor microenvironment. Here, histone lactylation drives ocular melanoma through the induction of YTH N6-Methyladenosine RNA Binding Protein 2 (YTHDF2) responsible for degrading N6-methylated RNA of the tumor supressors PER1 and TP53 (Yu et al., 2021). Histone lactylation indeed acts as a metabolic sensor. In non-small cell lung cancer, high intracellular lactate leads to the deposition of lactylation marks in the promoters of the glycolytic enzymes (Hexokinase 1 and pyruvate kinase-M), leading to a reduction in their expression, and a redirection of energy production from lactate-producing glycolysis to mitochondrial metabolism (Jiang et al., 2021). Furthermore, the activity of the histone acetylation reader BRD4, which is responsible for driving transcription of oncogenic programs such as those regulated by MYC, was found to depend on intracellular purine levels, thus balancing transcription with the availability of extracellular and salvaged purines (Li et al., 2021). As another example, the production of ketone bodies during fasting can induce histone (β-hydroxy) butyrylation, which in turn transcriptionally induces the starvation response (Xie et al., 2016). As elaborated on below, the epigenome as a metabolic sensor allows for rapid transcriptional adaptations to balance metabolism with cellular output.
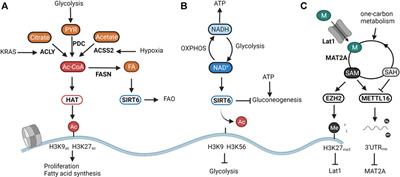
Besides the intracellular levels of acetyl-CoA, histone acetylation is further finetuned by deacetylation. A handful of studies have shown the ketone body d-β-hydroxybutyrate (βOHB) (Shimazu et al., 2013), lactate (Latham et al., 2012), and CoA derivatives (Vogelauer et al., 2012) to inhibit the activity of the classical histone deacetylases (HDAC1-10). Yet it is not known whether these act as metabolic sensors in physiological conditions. More evidence is available for highlighted the NAD+-dependent sirtuins (SIRT1-7)as textbook examples of metabolic sensors. NAD+ is a central metabolite that facilitates electron transfer derived from catabolic pathways. The ratio between NAD+ and its reduced form NADH thus represents cellular catabolic capacities and metabolic fitness. Whereas high NAD+ levels indicate low catabolic activity, low NAD+/NADH induces a shutdown of catabolism and regeneration of NAD Cantó et al., 2015. From all sirtuins, SIRT6 and SIRT7 are exclusively located in the nucleus. Whereas the poorly studied SIRT7 has been linked to DNA repair through deacetylation of H3K18ac (Barber et al., 2012), and to the activation of rRNA transcription through modulation of RNA polI (Ford et al., 2006), SIRT6 is an epigenetic regulator of glucose and fat metabolism through the removal of H3K9 and H3K56 acetylation (Chang et al., 2020). A shown in Figure 1B, when NAD+ levels are high, SIRT6 deacetylates H3K9 on the promoters of key glycolytic genes. This represses HIF-1α-dependent induction of glycolysis, and most importantly, of lactate dehydrogenase (LDH) (Zhong et al., 2010). In proliferating cancer cells, lactate secretion is responsible for regenerating NAD+ needed to maintain the glycolytic flux fueling cellular proliferation (Warburg effect) (Luengo et al., 2021). In normal cells, SIRT6 balances NAD+ levels by diverting glucose-derived carbons towards the mitochondrial TCA for the conversion of NAD+ to NADH and subsequent ATP production. Reduction of SIRT6 activity through low NAD+ availability or in an oncogenic setting results in a derepression of glycolytic genes, and, in the latter case, the acquisition of a metabolic profile driving uncontrolled proliferation (Sebastián et al., 2012; Choi et al., 2021). Especially in the liver, SIRT6 activity also determines the levels of gluconeogenesis. SIRT6 deacetylates and activates the acetyltransferase GCN5, resulting in an inhibition of PGC-1α-dependent gluconeogenesis. Furthermore, SIRT6 also deacetylates and induces nuclear exclusion of the gluconeogenic driver forkhead box protein O1 (FOXO1). As both reactions are dependent on NAD+, SIRT6 links gluconeogenic output and hepatic energy state through the NAD+/NADH ratio (Dominy et al., 2012; Zhang et al., 2014). Interestingly, the activity of SIRT6 was found to be amplified upon binding with free fatty acids (Feldman et al., 2013). As SIRT6 can also induce a switch from fatty acid synthesis to β-oxidation (FAO), its nuclear activity balances both glucose and fatty acid metabolism to NAD+ and fatty acid availability (Kim et al., 2010; Naiman et al., 2019) (Figure 1B).
etc, etc. I'll stop here. I started trying to quote stand-out sentences or paragraphs, until I realized I was just quoting the entire paper.
Yet, this interaction can also be the cause of disease. For example, a reduction in TET activity in hypoxic tumors leads to tumor evolution through the hypermethylation of tumor suppressors (Thienpont et al., 2016). Furthermore, methionine starvation within tumors inhibits the deposition of H3K79me2, resulting in low expression of STAT5 and immune dysfunction (Bian et al., 2020). Nevertheless, metabolic pathways also represent ideal targets to treat disease and epigenetic dysfunction. Dietary interventions or modulation of the intestinal microbiota can lead to a better outcome when combined with additional therapy (Iida et al., 2013; Gao et al., 2019; Parkhitko et al., 2019; Zheng et al., 2020).
To fully take advantage of metabolism as a therapeutical target, it is of extreme importance to understand how the interconnection between metabolism and epigenetics is regulated on a molecular level. Metabolic interventions lead to changes in epigenetic mark deposition. Furthermore, given that the Km values of epigenetic modifiers sometimes differ by several orders of magnitude, the intrinsic activity of chromatin modifiers might explain the specificity of marker deposition in different metabolic states (Reid et al., 2017). Yet, the numerous examples of nuclear-translocated metabolic enzymes demonstrate that molecular mechanisms exist to translate cytoplasmic cues into a nuclear metabolism. Especially for metabolic transformations, location matters and epigenetic modifiers probably respond more to local fluctuations in metabolite levels than those observed in other organelles Future studies should thus be focused on understanding subcellular metabolism and metabolic compartmentalization. This would encompass uncovering the regulation of nuclear translocation and investigating the subnuclear activity of metabolic enzymes and their operation in phase-separated liquid-liquid droplets. Interestingly, the activity and stability of transcription factors, generally regarded as the master regulators of expression, are also highly influenced by post-translational modifications (Benayoun and Veitia, 2009). The possibility thus arises that, as for the epigenome, also the local activity of transcription factors is determined through resident metabolic enzymes. Metabolic enzymes determine cell fate and transcription through epigenetic and transcription factor modification. Yet, it remains to be seen whether the location of metabolic enzymes on DNA matters. The key in understanding the interaction between metabolism and epigenetic modifications thus lies in studying their location.
@haidut
Frontiers | Metabolic Fuel for Epigenetic: Nuclear Production Meets Local Consumption
<p>Epigenetic modifications are responsible for finetuning gene expression profiles to the needs of cells, tissues, and organisms. To rapidly respond to envi...
www.frontiersin.org
Metabolic Fuel for Epigenetic: Nuclear Production Meets Local Consumption
- 1The Massachusetts General Hospital Cancer Center, Harvard Medical School, Boston, MA, United States
- 2The Broad Institute of Harvard and MIT, Cambridge, MA, United States
- 3Laboratory for Functional Epigenetics, Department of Human Genetics, KU Leuven, Leuven, Belgium
Introduction
In the last 2 decades, the field of epigenomics has identified an immense variety of chromatin modifiers that collaborate to establish the epigenetic code. Evidently, the deposition of epigenetic marks is determined through genetic regulation of the modifiers (Cavalli and Heard, 2019; Zhao et al., 2021). However, cellular metabolism has now emerged as a second layer of regulation. Indeed, whereas chromatin writers, readers, and erasers establish the marks, their activity critically depends on a handful of metabolites. In this way, chromatin modifiers act as sensors for metabolite levels and directly link mark deposition or removal with the metabolic state of the cell, organ, or organism (Boon et al., 2020a; Haws et al., 2020a; Dai et al., 2020).Cellular metabolism is the cluster of enzymatic reactions that occur in cells to transform nutrients such as glucose, fatty acids, amino acids, and vitamins into cellular components, energy, and reducing power. As metabolites can freely diffuse through the relatively large nuclear pores, the nucleus has been proposed to be in metabolic homeostasis with the cytoplasm (Wente and Rout, 2010; Cambronne et al., 2016). Yet, fluctuations in cytoplasmic metabolites should not lead to random epigenetic changes in the nucleus. Furthermore, both the cytoplasm and nucleus represent highly crowded environments where molecular bindings and collisions severely limit the diffusion of molecules (Ellis, 2001; Richter et al., 2008). It is therefore unlikely that passive diffusion of metabolites from the cytoplasm into the nucleus forms the basis of the metabolism-epigenetic link.
In this review, I explore where the metabolic fuel of epigenetic modifiers might be produced. First, I show a correlation between epigenetic marks and cytoplasmic metabolism. Second, I review studies indicating a distinct nuclear metabolism based on the import of metabolic enzymes. Last, I elaborate on observations indicating further subnuclear compartmentalization based on protein complexation and phase separation.
Diet is therefore increasingly appreciated as an additional way to target the epigenome to influence inflammation (Wang et al., 2018), metabolic disease (Yuan et al., 2018), and neurogenesis defects (Benjamin et al., 2017).
Cellular metabolism is not only regulated by cell-intrinsic signaling pathways. Given that metabolic pathways consume nutrients, there is extensive cross-reactivity between cellular metabolism and the composition of the nutrient microenvironment (Elia and Haigis, 2021). For example, in the pyruvate-and proline-rich lung microenvironment, metastatic breast cancer cells preferentially catabolise these nutrients to sustain energy production and the production of extracellular matrix components (Elia et al., 2017; Elia et al., 2019). Additionally, lactate buildup in metabolically active and poorly vascularized non-small-cell lung cancers (NSCLCs), induces the use of this “waste” product as a substantial carbon source for fueling mitochondrial oxidative metabolism (Faubert et al., 2017). Last, nutrient depletion observed in most tumors leads to metabolic inactivation of immune cells. T cells are metabolically outcompeted by tumor cells in terms of glucose (Cascone et al., 2018), methionine (Bian et al., 2020), and arginine catabolism (Geiger et al., 2016).
The epigenome thus lies between the nutrient microenvironment and the regulation of cell fate. In this way, epigenetics might act as a metabolic antenna that probes cellular metabolism and translates environmental changes into transcriptional responses. Although the exact modifier was not identified, recently, a novel histone modification, histone lactylation, was indeed found to modulate wound healing and homeostasis of macrophages as a response to high levels of lactate as found in sepsis and hypoxia (Zhang et al., 2019). These findings also translate to the lactate-high tumor microenvironment. Here, histone lactylation drives ocular melanoma through the induction of YTH N6-Methyladenosine RNA Binding Protein 2 (YTHDF2) responsible for degrading N6-methylated RNA of the tumor supressors PER1 and TP53 (Yu et al., 2021). Histone lactylation indeed acts as a metabolic sensor. In non-small cell lung cancer, high intracellular lactate leads to the deposition of lactylation marks in the promoters of the glycolytic enzymes (Hexokinase 1 and pyruvate kinase-M), leading to a reduction in their expression, and a redirection of energy production from lactate-producing glycolysis to mitochondrial metabolism (Jiang et al., 2021). Furthermore, the activity of the histone acetylation reader BRD4, which is responsible for driving transcription of oncogenic programs such as those regulated by MYC, was found to depend on intracellular purine levels, thus balancing transcription with the availability of extracellular and salvaged purines (Li et al., 2021). As another example, the production of ketone bodies during fasting can induce histone (β-hydroxy) butyrylation, which in turn transcriptionally induces the starvation response (Xie et al., 2016). As elaborated on below, the epigenome as a metabolic sensor allows for rapid transcriptional adaptations to balance metabolism with cellular output.
Besides the intracellular levels of acetyl-CoA, histone acetylation is further finetuned by deacetylation. A handful of studies have shown the ketone body d-β-hydroxybutyrate (βOHB) (Shimazu et al., 2013), lactate (Latham et al., 2012), and CoA derivatives (Vogelauer et al., 2012) to inhibit the activity of the classical histone deacetylases (HDAC1-10). Yet it is not known whether these act as metabolic sensors in physiological conditions. More evidence is available for highlighted the NAD+-dependent sirtuins (SIRT1-7)as textbook examples of metabolic sensors. NAD+ is a central metabolite that facilitates electron transfer derived from catabolic pathways. The ratio between NAD+ and its reduced form NADH thus represents cellular catabolic capacities and metabolic fitness. Whereas high NAD+ levels indicate low catabolic activity, low NAD+/NADH induces a shutdown of catabolism and regeneration of NAD Cantó et al., 2015. From all sirtuins, SIRT6 and SIRT7 are exclusively located in the nucleus. Whereas the poorly studied SIRT7 has been linked to DNA repair through deacetylation of H3K18ac (Barber et al., 2012), and to the activation of rRNA transcription through modulation of RNA polI (Ford et al., 2006), SIRT6 is an epigenetic regulator of glucose and fat metabolism through the removal of H3K9 and H3K56 acetylation (Chang et al., 2020). A shown in Figure 1B, when NAD+ levels are high, SIRT6 deacetylates H3K9 on the promoters of key glycolytic genes. This represses HIF-1α-dependent induction of glycolysis, and most importantly, of lactate dehydrogenase (LDH) (Zhong et al., 2010). In proliferating cancer cells, lactate secretion is responsible for regenerating NAD+ needed to maintain the glycolytic flux fueling cellular proliferation (Warburg effect) (Luengo et al., 2021). In normal cells, SIRT6 balances NAD+ levels by diverting glucose-derived carbons towards the mitochondrial TCA for the conversion of NAD+ to NADH and subsequent ATP production. Reduction of SIRT6 activity through low NAD+ availability or in an oncogenic setting results in a derepression of glycolytic genes, and, in the latter case, the acquisition of a metabolic profile driving uncontrolled proliferation (Sebastián et al., 2012; Choi et al., 2021). Especially in the liver, SIRT6 activity also determines the levels of gluconeogenesis. SIRT6 deacetylates and activates the acetyltransferase GCN5, resulting in an inhibition of PGC-1α-dependent gluconeogenesis. Furthermore, SIRT6 also deacetylates and induces nuclear exclusion of the gluconeogenic driver forkhead box protein O1 (FOXO1). As both reactions are dependent on NAD+, SIRT6 links gluconeogenic output and hepatic energy state through the NAD+/NADH ratio (Dominy et al., 2012; Zhang et al., 2014). Interestingly, the activity of SIRT6 was found to be amplified upon binding with free fatty acids (Feldman et al., 2013). As SIRT6 can also induce a switch from fatty acid synthesis to β-oxidation (FAO), its nuclear activity balances both glucose and fatty acid metabolism to NAD+ and fatty acid availability (Kim et al., 2010; Naiman et al., 2019) (Figure 1B).
etc, etc. I'll stop here. I started trying to quote stand-out sentences or paragraphs, until I realized I was just quoting the entire paper.
Concluding Remarks
The field of epigenomics has evolved tremendously from the first descriptions of DNA methylation changes during developing embryos. Although the image of epigenetic marks as static guardians of cellular identity as culminated in Waddington’s epigenomic landscape is, of course, valid, the epigenome also represents a much more dynamic manner of regulating transcription (Allis and Jenuwein, 2016). As epigenetic modifiers are enzymes, they can probe the cellular metabolome and sense changes in levels of metabolites and vitamins. As described, this mechanism allows for balancing metabolite levels through rapid transcriptional changes. Inputs into the transcriptional regulation through the metabolic epigenome include nutritional status (Boon et al., 2020a) (including oxygen availability Thienpont et al., 2016 and pH McBrian et al., 2013), signaling pathways (Carrer et al., 2019), circadian oscillations (Masri and Sassone-Corsi, 2013; Greco et al., 2020).Yet, this interaction can also be the cause of disease. For example, a reduction in TET activity in hypoxic tumors leads to tumor evolution through the hypermethylation of tumor suppressors (Thienpont et al., 2016). Furthermore, methionine starvation within tumors inhibits the deposition of H3K79me2, resulting in low expression of STAT5 and immune dysfunction (Bian et al., 2020). Nevertheless, metabolic pathways also represent ideal targets to treat disease and epigenetic dysfunction. Dietary interventions or modulation of the intestinal microbiota can lead to a better outcome when combined with additional therapy (Iida et al., 2013; Gao et al., 2019; Parkhitko et al., 2019; Zheng et al., 2020).
To fully take advantage of metabolism as a therapeutical target, it is of extreme importance to understand how the interconnection between metabolism and epigenetics is regulated on a molecular level. Metabolic interventions lead to changes in epigenetic mark deposition. Furthermore, given that the Km values of epigenetic modifiers sometimes differ by several orders of magnitude, the intrinsic activity of chromatin modifiers might explain the specificity of marker deposition in different metabolic states (Reid et al., 2017). Yet, the numerous examples of nuclear-translocated metabolic enzymes demonstrate that molecular mechanisms exist to translate cytoplasmic cues into a nuclear metabolism. Especially for metabolic transformations, location matters and epigenetic modifiers probably respond more to local fluctuations in metabolite levels than those observed in other organelles Future studies should thus be focused on understanding subcellular metabolism and metabolic compartmentalization. This would encompass uncovering the regulation of nuclear translocation and investigating the subnuclear activity of metabolic enzymes and their operation in phase-separated liquid-liquid droplets. Interestingly, the activity and stability of transcription factors, generally regarded as the master regulators of expression, are also highly influenced by post-translational modifications (Benayoun and Veitia, 2009). The possibility thus arises that, as for the epigenome, also the local activity of transcription factors is determined through resident metabolic enzymes. Metabolic enzymes determine cell fate and transcription through epigenetic and transcription factor modification. Yet, it remains to be seen whether the location of metabolic enzymes on DNA matters. The key in understanding the interaction between metabolism and epigenetic modifications thus lies in studying their location.
Last edited: