md_a
Member
- Joined
- Aug 31, 2015
- Messages
- 468
Discussion
This study demonstrated that chronic VDD resulted in lung fibrosis, partly through the activation of the renin-angiotensin system. The AT1 blocker losartan and the renin blocker Aliskiren could alleviate lung fibrosis induced by chronic VDD. This effect was independent of the regulatory role of RAS on blood pressure.Generally there are three types of VDD models: prenatal, postnatal and whole-life VDD. VDD is exclusively achieved through diet and light control, as was done in our study. By using a whole-life VDD model, one study found that VDD causes deficits in lung function that cannot be explained by somatic growth and lung volume1. A later study on pre- and postnatal VDD found that prenatal VDD caused diminished tracheal diameter, tracheal cartilage thickness and alveolar simplification which led to increased airway resistance and decreased pulmonary compliance, while postnatal VDD had little influence on tracheal morphology and lung function15. Postnatal vitamin D supplementation could improve lung function but not normalize tracheal abnormalities. Another similar study found that the increase in inflammatory molecules, airway smooth muscle mass and baseline airway resistance were caused largely by VDD in utero and postnatal supplementation can do nothing but relieve airway hyperresponsiveness16. The changes, however, mainly happen in the latter stage of alveolar development because comparisons between VDD and normal subjects did not show significant difference in protein expressions at E14.5 and E17.517.
Previous studies on VDD exclusively focused on its aggravating effect on pre-existing lung diseases that subsequently led to lung fibrosis. Those diseases are mostly chronic, including bronchopulmonary dysplasia, asthma, cystic fibrosis, COPD and interstitial lung disease18, 19. Bronchopulmonary dysplasia attacks premature births and proceeds into childhood. Asthma generally starts in childhood or early adulthood, while others attack mostly adults and the aged. This study, however, aimed to explore the pro-fibrotic role of VDD itself. Since these diseases cover almost the entire span from in uterus to old age and fibrosis may happen any time in this span, we used a whole-life VDD model to stay in synchronism with the process of the diseases.
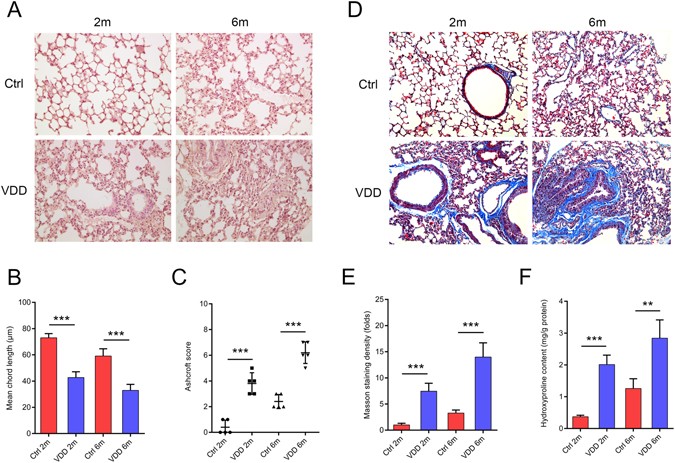
The role of vitamin D in tissue fibrosis has been demonstrated in many organs. Vitamin D analogues were curative in fibrosis models of the kidney5, peritoneum6, liver7 and lung3. VDD worsened 2,4,6-trinitrobenzene sulfonic acid-induced intestinal fibrosis in mice20. Serum TGF-β levels were inversely correlated with the vitamin D levels in patients with bone marrow fibrosis21. VDD has been especially associated with lung fibrosis. It has been shown to increase pulmonary exacerbations and decrease lung function in patients with cystic fibrosis12. However, the question of whether vitamin D could aggravate pre-existing fibrosis or induce fibrosis itself remains unclear. In chronic vitamin D deficient mice, alveolar simplification and stunted lung development were present right after birth (see Supplementary Figure 3). Two months after birth we observed pulmonary fibrosis, the accumulation of collagen and the disruption of normal structures. Increased collagen secretion causes stiffness of the tissue and a loss of elasticity. Uncontrolled myofibroblast proliferation, represented by increased α-SMA levels, adds to the deposition of ECM. In this way, the alveolar structure is destroyed, leading to a reduced mean free distance of the air space. These results showed that chronic VDD could lead to lung fibrosis. This pathological change would aggravate with prolonged VDD.
Vitamin D and RAS are so closely related that hypovitaminosis D has been described as the other face of RAS activation22. Vitamin D receptor (VDR) knockout mice showed an over-expression of renin, which transformed more angiotensinogen into angiotensin II, so that the animals would increase water intake and intestinal salt absorption13. Vitamin D could reduce the abnormal increase in RAS in pancreatic islets23. By activating VDR, vitamin D reduces apoptosis through a blockade of RAS in morphine-induced T cell apoptosis24. Chronic RAS activation has been shown to promote lung fibrosis, causing an increased expression of ECM proteins and fibrogenetic factors as well as decreased pulmonary compliance14.
Because the activation of RAS has been reported to induce lung fibrosis both in transgenic animals and in disease models14, 25, 26, it was reasonable to hypothesize that the lung fibrosis observed in vitamin D deficient mice is associated with the increased expression of RAS components. To clarify this, we administered RAS antagonists to the vitamin D deficient mice. Losartan is a blocker of AT1R, which prohibits angiotensin II from binding to AT1R.Therefore the expression of renin is increased due to a negative feedback. Aliskiren blocks the production of renin. Decreased renin up-regulates the expression of angiotensinogen also through a negative feedback. The renin antagonist aliskiren and the AT1R blocker losartan could significantly rescue the tissue damage and alleviate lung fibrosis. This protective effect of RAS blockers has also been reported on lacrimal gland fibrosis in chronic graft-versus-host disease, as well as in bleomycin-induced and radiation-induced lung fibrosis27,28,29.
However, in our study, the influence of RAS may be confounded by its regulatory effect on blood pressure, because hypertension is an independent risk factor of lung fibrosis and may be induced by RAS activation. Thus, we gave hydralazine to the vitamin D deficient mice as a control, which could restore normal blood pressure without interfering with RAS. Hydralazine did not show any preservative effect, indicating that the induction of lung fibrosis by RAS was not due to hypertension.
In chronic vitamin D deficient mice, the expression of renin induces more angiotensinogen to be transformed into angiotensin II, which, by combining with the angiotensin type 1 receptor, activates TGF-β1. TGF-β1 plays a vital role in preserving ECM, inducing fibroblast proliferation and transforming fibroblasts into myofibroblasts24. Activated fibroblasts deposit collagen I, an important component of ECM, which causes the thickening of the alveolar walls30. Increased numbers of myofibroblasts secrete more α-SMA, as observed in our study. Meanwhile, TGF-β1 stimulates the expression of angiotensinogen and the angiotensin type 1 receptor, forming the angiotensin-TGF-β1 crosstalk30. This crosstalk further enhances the profibrotic effect of angiotensin.
Still other mechanisms might explain the role of VDD in lung fibrosis, including the nuclear factor-κB and wnt/β-catenin pathways8, and the failure to address to them is one limitation of this study. Moreover, we did not testify our findings in the human population. Large-scale population-based studies are still needed to confirm our conclusions.
In summary, chronic VDD may induce over-activation of RAS, which subsequently stimulates the expression of TGF-β1 and activates the fibrotic cascade. Structural damage and ECM deposition aggravate with prolonged deficiency. This effect of RAS is independent of the classical function of blood pressure regulation. Our study draws attention to chronic VDD in the population, which might be a cause of lung fibrosis.
Chronic vitamin D deficiency induces lung fibrosis through activation of the renin-angiotensin system - Scientific Reports
Pulmonary fibrosis, which influences lung function and exacerbates a patient’s condition, is the ultimate stage of many lung diseases. Vitamin D deficiency is associated with pulmonary fibrosis and impaired lung function, but the underlying mechanism has not yet been fully elucidated. Moreover...
 www.nature.com
www.nature.com

